Частота регистрации нейропатических нарушений при сахарном диабете (СД) колеблется от 15 до 90%, что обусловлено принципиальными различиями в группах обследуемых пациентов и диагностических подходах. Так, по данным Oxford Community Diabetes Study, распространенность диабетической нейропатии (ДН) при СД типа 1 с 10-летним «стажем» заболевания составляет 20,9%, а при СД типа 2 — 5,8%. D. Ziegler et al. сообщили о большей частоте регистрации ДН при СД типа 2 (34,3% против 25,3% при СД типа 1) [63, 66, 72].
В то же время общеизвестно, что ДН в большинстве случаев является причиной ампутации конечностей и, соответственно, инвалидизации. По данным International Working Group on the Diabetic Foot [21], от 40 до 70% всех ампутаций нижних конечностей связано с сахарным диабетом, в 85% случаев этим ампутациям предшествуют язвенные дефекты.
Целью настоящей публикации не является подробный анализ патогенеза, классификационных подходов и принципов диагностики ДН, поэтому напомним лишь об основных патофизиологических моментах, лежащих в основе развития патологического процесса (рис.1).
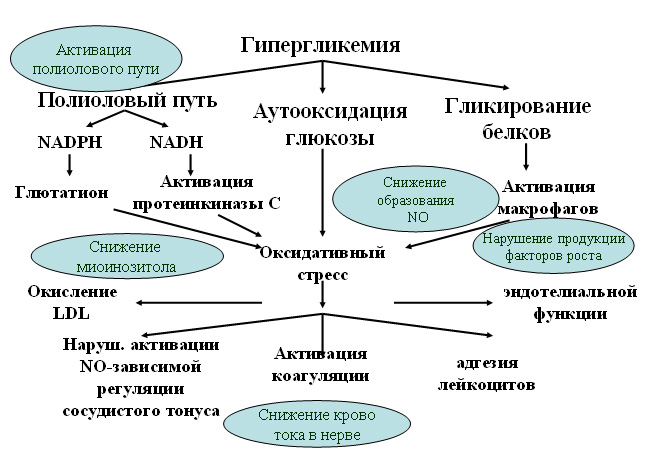
Рис.1. Основы патогенеза ДН
В патогенезе диабетической нейропатии пусковым звеном является гипергликемия с последующим нарушением метаболизма в нейронах с развитием их отека и дисфункции (рис.1) вследствие:
• активации альдозоредуктазы (ключевого фермента полиолового пути метаболизма глюкозы) и накопления гидрофильного сорбита и фруктозы;
• активации протеинкиназы С с развитием дефицита мио-инозитола, токсического воздействия свободных радикалов на нейроны, а также активированием и фосфорилированием цитозольной фосфолипазы А2 (сPLA2) с увеличением образования арахидоната и простагландина Е2, участвующих в изменении проницаемости сосудистой стенки и тромбообразовании;
• усиленного образования действующих вазоконстрикторно простагландинов, которые противостоят NO-зависимой вазодилатации;
• прямого глюкотоксического стресса и неферментативного гликирования белков с формированием конечных продуктов гликозилирования, активацией фактора роста фибробластов и нарушением функции коллагена I и IV типов, липопротеинов, иммуно-глобулина G, ламинина, гепаран сульфата, протеогликанов;
• микроангиопатического поражения vasa nervorum и развития хронической эндоневральной ишемии;
• акселерации атеросклероза и др. [1, 4, 28, 32, 36, 41, 56, 57].
Cогласно консенсусу по диабетической нейропатии (1994), среди патофизиологических механизмов выделены четыре основных, которые определяют патогенетические принципы лечения [3]:
• образование конечных продуктов гликозилирования;
• нарушение микроциркуляции;
• увеличение образования свободных радикалов;
• ослабление антиоксидантной защиты.
Кроме указанных патофизиологических механизмов следует акцентировать внимание на основных симптомах поражения нервной системы при СД, находящихся в зависимости от поражения периферической (автономная и дистальная ДН) или центральной нервной системы. При дистальной ДН, которая диагностируется наиболее часто, клинические проявления включают парестезии (онемение, покалывание, жжение, чувство «ползания мурашек» и др.), судороги, нарушение рефлексов, болевой синдром, миопатический синдром и, возможно, трофические язвы.
До последнего времени возможность развития диабетической энцефалопатии трактовалась неоднозначно, что обусловлено сложностями в унификации диагностических подходов. В настоящее время единые подходы по-прежнему отсутствуют, однако наличие «центральной» формы нейропатии признано доказанным. Клинические проявления энцефалопатии включают интеллектуально-мнестические нарушения, головные боли, нарушения настроения [55, 63].
Несмотря на успехи в понимании патогенеза поражения нервов при СД, до сих пор не разработан метод лечения, который стал бы стандартом терапии ДН и был бы утвержден международными организациями [9, 21, 25].
Лечение периферической ДН принято разделять на патогенетическое и симптоматическое [61]. Патогенетическая терапия включает:
• мероприятия, направленные на достижение и поддержание стойкой компенсации СД;
• ингибиторы альдозоредуктазы — блокаторы полиолового пути метаболизма глюкозы;
• витамины группы В — бенфотиамин и цианокобаламин – ингибиторы гликолиза, блокирующие глюкотоксический эффект и формирование конечных продуктов гликозилирования;
•? α-липоевая кислота — активирует митохондриальные ферменты и окисление глюкозы, тормозит глюконеогенез;
• эссенциальные жирные кислоты — обладают антиоксидантным эффектом и снижают гиперлипидемию.
Одна из целей симптоматической терапии — устранение болевого синдрома, который часто ассоциирован с нарушениями сна и существенно снижает качество жизни при СД. Кроме того, симптоматическая терапия включает мероприятия, направленные на:
• устранение судорог в конечностях;
• профилактику и лечение язвенных дефектов стопы;
• коррекцию минеральной плотности костной ткани при развитии остеопороза;
• лечение сопутствующих инфекций и т.д.
Патогенетическая терапия
Роль декомпенсации СД в патогенезе развития и прогрессии различных форм ДН (дистальной и автономной) доказана во всех крупных исследованиях (Diabetes Control Complication Study, United Kingdom Prospective Diabetes Study), поэтому прин-ципы лечения и целевые значения компенсации СД не отличаются от общепринятых (рис. 2) и включают использование бигуанидов (метформина), препаратов сульфонилмочевины, тиазолидиндионов и инсулина. Однако при выраженном болевом синдроме или наличии язвенных дефектов оптимальной является инсулинотерапия (при СД типа 2 возможно и даже предпочтительно комбинированное лечение оральными препаратами и инсулином) [20, 45, 60, 62].
.jpg)
Рис. 2. Цель гликемического контроля
Ингибиторы альдозоредуктазы блокируют накопление сорбитола, фруктозы и снижение миоинозитола в нервных окончаниях, что подтверждено оптимизацией нервной проводимости, особенно в эксперименте. Препараты, используемые в клинической практике (Statil, Sorbinil, Alredase, Tolrestat, Ponalrestat, Zenarestat, Zopolrestat), по результатам клинических исследований продемонстрировали минимальный эффект при манифестной нейропатии и значимый превентивный эффект в отношении прогрессии поражения нервов [49]. В то же время длительный прием ингибиторов альдозоредуктазы (Zopolrestat) (EASD, 2007) не приводит к снижению риска ампутаций, восстановлению чувствительности, уменьшению симптомов ДН, улучшению качества жизни [46]. Патогенетическая роль полиолового пути в развитии ДН инициировала исследования по оценке эффективности ингибирования другого фермента – сорбит-дегидрогеназы. Для восполнения дефицита миоинозитола рассматривается вопрос о возможности использования специальных пищевых добавок, содержащих миоинозитол.
Поскольку диагностика доклинических стадий ДН затруднена и, как правило, проводится с запозданием, после появления клинических признаков заболевания, применение ингибиторов альдозоредуктазы в патогенетической терапии ограничено [5, 59, 63].
Витамины группы В оказывают метаболическое влияние на аксональный транспорт и процессы миелинизации в периферических нервных волокнах. Воздействие витаминов группы В, в первую очередь тиамина, на центральную нервную систему опосредовано через метаболизм ГАМК и серотонина и вызывает анальгетический эффект.
Эти эффекты возможны только при достижении высокой концентрации витамина в нервной ткани, чего можно добиться при использовании либо высоких дозировок, либо специально разработанных жирорастворимых форм витамина В1 (бенфотиамин) [16, 27]. Препаратами выбора являются нейромультивит, мильгамма, мильгамма N (табл. 1). Результаты их применения свидетельствуют о снижении болевых ощущений, улучшении вибрационной чувствительности и возрастании скорости нейронального проведения через 3—6 недель от начала лечения [58]. Например, исследование BEDIP (Benfotiamine in the Treatment of Diabetic Polyneuropathy study) доказало эффективность использования бенфотиамина в течение 3 недель при ДН: отмечено снижение счета по NDS (Neuropathy Disability Score), улучшение вибрационной чувствительности и «общего состояния» по сравнению с плацебо [29, 31]. К сожалению, исследования, свидетельствующие о долгосрочной эффективности терапии препаратами витаминов группы В, отсутствуют, что не позволяет экспертам внести их в международные рекомендации.
Таблица 1. Препараты витаминов группы В, используемые для лечения диабетической нейропатии
|
Название препарата
|
Состав
|
Форма выпуска
|
Дозировка и длительность лечения
|
|
Нейромультивит
|
Тиамин — 100 мг, пиридоксин — 200 мг, цианокобаламин — 200 мкг
|
Таблетки
|
По 1 табл. 2—3 раза в день 4—6 недель
|
|
Мильгамма
|
Бенфотиамин –100 мг, пиридоксин – 100 мг
|
Ампулы
|
Внутримышечно 1 раз в день в течение 4—6 недель
|
|
Мильгамма N
|
Тиамин — 50 мг, пиридоксин — 50 мг, цианокобаламин — 500 мкг
|
Ампулы, драже
|
Внутримышечно 1 раз в день в течение 5—10 дней, затем 2—3 драже в день в течение 4—6 недель
|
|
Бенфогамма
|
Бенфотиамин — 15 мг
|
Драже
|
1—2 драже ежедневно в течение 4—6 недель
|
Альфа-липоевая (тиоктовая) кислота является коэнзимом, который выступает биокатализатором, активизирующим митохондриальные ферменты, тормозящим глюконеогенез и кетогенез, вызывающим антиоксидантный эффект, блокирующим образование свободных радикалов, снижающим синтез холестерина, замедляющим гликирование протеинов и формирование конечного продукта гликозилирования, предотвращающим потерю миоинозитола, улучшающим чувствительность к инсулину и способствующим утилизации глюкозы тканями. В результате улучшается эндоневральный кровоток, различные виды чувствительности, возрастает скорость проведения нервного импульса и уменьшаются моторные расстройства.
Доказано, что альфа-липоевая кислота оказывает влияние на параметры оксидативного стресса (NO, конечный продукт гликозилирования, ядерный фактор каппа (NF -kB) и др.), что позволило планировать эксперимент по оценке ее воздействия на развитие ретинопатии [17, 47, 48, 52]. В результате при морфометрии сетчатки отмечено снижение выраженности микрососудистых нарушений [38]. Полученные результаты свидетельствуют о позитивной протективной роли альфа-липоевой кислоты в развитии ретинопатии [37]. Подробнее читайте на http://diabet-med.com/alfa-lipoevaya-kislota/. Существуют различные соли альфа-липоевой кислоты: этилендиаминовая, трометамоловая и меглюминовая. Наиболее часто используется трометамоловая соль, известная как Тиоктацид [35].
В настоящее время завершены многочисленные рандомизированные двойные слепые плацебо-контролируемые исследования эффективности альфа-липоевой кислоты. Результаты приведены в табл. 2.
Таблица 2. Рандомизированные двойные слепые плацебо-контролируемые исследования по эффективности тиоктовой кислоты*
|
Исследование
|
N
|
Введение/доза ( мг)
|
Длительность
|
Результаты
|
Безопасность
|
|
ALADIN I
|
382
|
в/в, 100/600/1200 плацебо
|
3 нед.
|
TSS+, NDS+, HPAL+
|
Хорошая
|
|
ALADIN II
|
65
|
600/1200/плацебо
|
2 года
|
NCV suralis+, NCV tibialis +, SNAP+, DML-, NDS-
|
Хорошая
|
|
ALADIN III
|
508
|
в/в 600, перорально 1200/плацебо
|
3 нед. в/в, 6 мес. Перорально
|
TSS+, Y Aladin I HPAL+, NDS+, NDSLL+
|
Хорошая
|
|
DEKAN
|
73
|
Перорально 800/плацебо
|
4 мес
|
HRV+, QT-
|
Хорошая
|
|
ORPIL
|
24
|
Перорально 1800/плацебо
|
3 нед.
|
TSS+, HPAL+, NDS+
|
Хорошая
|
П р и м е ч а н и е: оценочные критерии: TSS—Total Symptom Score; NDS—Neuropathy Disability Score; HPAL—Hamburg Pain Adjective List; SNAP – Sensory Nerve Action Potential; NCV — Nerve Conduction Velocity; DML — Distal Motor Latency; (+) – улучшение, (-) – нет эффекта. * Исследования выполнены с применением препарата «Тиоктацид».
В исследовании ALADIN I (Alpha-lipoic acid in Diabetic Neuropathy I) определена оптимальная терапевтическая доза альфа-липоевой кислоты — 600 мг внутривенно (эффект меньшей дозы (100 мг) сравним с эффектом плацебо) и установлено снижение болевых ощущений, чувства жжения, онемения. В другом исследовании (ALADIN II) доказано, что оральный прием тиоктацида в дозе 600 или 1200 мг в течение 2 лет (после пятидневного периода насыщения внутривенным введением) улучшает функцию нерва, увеличивая скорость проведения нервного импульса. При этом 89% пациентов в группе, получавшей 600 мг, и 94% в группе, получавшей 1200 мг альфа-липоевой кислоты в течение 2 лет, оценили переносимость препарата как хорошую и очень хорошую. Авторы сделали вывод о том, что переносимость препарата при длительном приеме сравнима с плацебо [53, 68 — 73].
Аналогичные результаты имели место в исследовании SYDNEY, в котором по шкалам TSS, NDS и при нейромиографии отмечено уменьшение характерных нейропатических симптомов с высокой степенью достоверности [10].
При оценке альфа-липоевой кислоты изучалось ее влияние не только на дистальную ДН, но и на течение автономной кардиальной ДН.
Препараты альфа-липоевой кислоты выпускаются как в инфузионной, так и в таблетированной форме (тиоктацид, берлитион, эспалипон, тиогамма и др.). Стандартный курс лечения начинают с инфузионного введения препарата в дозе 600 мг в сутки внутривенно капельно на 150,0 мл 0,9% раствора NaCl в течение 3 нед. (с перерывами в выходные дни) с последующим пероральным приемом препарата в течение 2—3 мес по 600 мг/сут. Учитывая фармакокинетические особенности всасывания таблетированных форм альфа-липоевой кислоты в кишечнике, прием таблеток рекомендуется осуществлять не менее чем за 30 мин до приема пищи.
В настоящее время разработана особая форма — тиоктацид БВ, отличающаяся от стандартной добавлением вспомогательных компонентов к ядру таблетки и изменением пленочного покрытия, что обеспечило оптимизацию фармакокинетики препарата, улучшенную биодоступность и снижение коэффициента вариабельности уровня тиоктовой кислоты в плазме крови. С использованием тиоктацида БВ проведено исследование ORPIL (Oral Pilot), доказавшее равноценную эффективность препарата. При оценке результатов применяли три различных подхода (шкалы): TSS, NDS и HPAL, по которым отмечено не только снижение баллов TSS и NDS, но и уменьшение болевого стресса, тревожности, изменений интенсивности и ритма болей [72].
В результате предложена альтернативная схема, включающая начальную терапию по 600 мг альфа-липоевой кислоты 3 раза в день в течение 3 недель (1800 мг/сут) и поддерживающую терапию по 600 мг 1 раз в день утром натощак на протяжении 2–3 месяцев [73]. Для оценки эффективности указанных методик было инициировано многоцентровое исследование NATHAN (Neurological Assessment of Thiоctic Acid in Neuropathy study) I. В сентябре 2007 г. D. Ziegler были доложены результаты этого пролонгированного (192 нед.) исследования. Отмечена хорошая переносимость препарата и улучшение самочувствия в процессе лечения по сравнению с группой плацебо. В то же время не зарегистрировано снижения скорости прогрессии нейропатии, не наблюдалось улучшения показателей различных видов чувствительности и нервной проводимости [70].
Эссенциальные жирные кислоты класса омега-3 и омега-6 способствуют образованию эйкозаноидов, которые оказывают антиоксидантное и гиполиподемическое действие, снижают агрегацию тромбоцитов (эйколен, льняное масло, тыквеол и др.). Однако клинические исследования, подтверждающие их эффективность при ДН, не проводились [2, 18, 65].
Симптоматическая терапия
Анальгетическая терапия базируется на использовании принципиально различных патогенетических подходов и отсутствии значимого эффекта при назначении анальгина, парацетамола, нестероидных противовоспалительных препаратов [43, 50].
Сочетание ДН с депрессией дало повод использовать антидепрессанты (в виде монотерапии или в комбинации с транквилизаторами). Возможный механизм действия антидепрессантов заключается в угнетении обратного захвата норадреналина в синапсах центральной нервной системы. В течение многих лет отдавалось предпочтение иcпользованию трициклических антидепрессантов, в частности амитриптилину в дозе от 25 до 150 мг на ночь [22]. Показан дозозависимый эффект трициклических антидепрессантов в отношении жгучих и стреляющих болей у пациентов с депрессией и без нее, хотя эффективность лечения у больных с депрессией оказалась выше [42, 59].
В настоящее время оптимальный эффект при ДН получен при использовании дулоксетина (Cymbalta) [26, 51]. Дулоксетин — блокатор обратного захвата серотонина и норэпинефрина пресинаптической мембраной. Воздействие дулоксетина основано на блокаде на уровне головного и спинного мозга. В настоящее время завершены многочисленные двойные слепые рандомизированные исследования, подтверждающие эффективность дулоксетина при болевых формах ДН и депрессиях. Так, J. Raskin et al. доказали не только статистически достоверное снижение болевого синдрома по специальной 11-пунктовой шкале (Likert scale или Average Pain Score, согласно которой 0 — боли нет, 10 — самая сильная боль), но и хорошую переносимость дулоксетина при длительном применении (52 нед.) [51]. Такие же результаты получены при сравнении эффективности дулоксетина в различных дозах (20, 60 и 120 мг/сут) и плацебо при болевой форме ДН в течение 12 недель: по общей шкале боли (Average Pain Score) отмечено снижение на 50% при использовании дозы 120 мг/сут и на 20% — 60 мг/сут.
Возможно применение центральных миорелаксантов, однако доказательной базы относительно их более высокой эффективности при ДН нет. Центральные миорелаксанты — гетерогенная группа, включающая тизанидин (агонист альфа-2-адренорецепторов), баклофен (антагонист GABAB-рецепторов), диазепам (агонист GABAA-рецепторов), мемантин (ингибитор NMDA-зависимых каналов), толперизон (блокатор Na-каналов и стабилизатор мембран) [7]. С позиций формирования боли и сохранения качества жизни при спастическом синдроме имеет значение уменьшение степени выраженности спазма, улучшение кровообращения в мышце и, наконец, отсутствие мышечной слабости после приема лекарственного средства.
Препаратами выбора являются тиназидина гидрохлорид и толперизона гидрохлорид. Тиназидин (сирдалуд) — релаксант скелетной мускулатуры центрального действия. Стимулируя пресинаптические альфа-2-рецепторы, ингибирует выработку аминокислот, стимулирующих Н-метил-D-аспартатные (NMDA) рецепторы, блокирует на уровне промежуточных нейронов спинного мозга передачу возбуждения, подавляет избыточный мышечный тонус и вызывает обезболивающий эффект. Одновременно обладает центральным анальгезирующим действием. Назначается по 2—4 мг 3 раза в сутки (не более 36 мг/сут).
Толперизон (мидокалм) — миорелаксант центрального действия, действует посредством влияния на каудальную часть ретикулярной формации и подавляет патологическую спинномозговую рефлекторную активность, что подтверждено снижением суммарного показателя выраженности клинических симптомов при болях в спине или конечностях. Это является следствием устранения эффекта мышечного спазма, который сопровождается стимуляцией ноцирецепторов мышц, локальной ишемией и последующим выбросом медиаторов воспаления и боли (простагландинов, серотонина, брадикинина). Назначается толперизон по 50 (150) мг 3 раза в сутки или внутримышечно 100 мг 2 раза в сутки [11].
При мышечных судорогах в ногах могут быть назначены препараты магния, в том числе в комбинации с витамином В6 (пиридоксин). Дефицит магния сопровождается нарушением мышечной релаксации, уменьшением резервного пула калия и относительной гипокальциемией, что в конечном итоге приводит к возникновению мышечных судорог в отдельных мышцах или группах мышц. Препараты магния – магне В6, магвит, магнерот — назначают при сердечно-сосудистой патологии (инфаркте миокарда, недостаточности кровообращения, аритмиях, спазмах сосудов), а ДН часто развивается у пациентов с исходной кардиальной патологией.
Антиконвульсанты (карбамазепин по 100 мг 2 раза в день (до 400 мг 3 раза в день), фенитоин (по 1 табл. 2—3 раза в сутки)) также снижают болевые ощущения при ДН. В настоящее время разработан новый антиконвульсант для лечения диабетической нейропатии — лакозамид, который обеспечивает селективную медленную инактивацию калиевых каналов, что выгодно отличает его от других антиконвульсантов, способных воздействовать на различные типы рецепторов и модулировать ответ медиатора коллапсина (CRMP-2). Лакозамид в дозе 200—600 мг/сут снижает болевой синдром при ДН.
Внимания заслуживает группа противосудорожных препаратов (габапентин, прегабалин), являющихся группой выбора в случае необходимости купирования болевого синдрома при ДН. Действие габапентина основано на сходстве его молекулы с молекулой ГАМК (GABA), но при этом молекула габапентина не обладает ГАМК-эргическими свойствами, не влияет на захват и метаболизм ГАМК. Габапентин (нейронтин, тебантин) реализует свой эффект через блокаду ионов кальция, через кальций-зависимые каналы, снижает глутамат-зависимую гибель нейронов, увеличивает синтез ГАМК и подавляет высвобождение нейротрансмиттеров моноаминовой группы. Перечисленные особенности обусловливают отличия габапентина от других противосудорожных препаратов. Завершены и опубликованы результаты двойного слепого рандомизированного исследования по оценке влияния габапентина на выраженность боли при ДН в дозе 1800 мг/сут. Зарегистрировано снижение среднего недельного балла боли до 4,5 через 2 недели лечения и до 3,5 – через 8 недель [13, 14]. Одновременно фиксировали достоверное улучшение ночного сна и качества жизни (по опроснику SF -36) за счет уменьшения болевых телесных ощущений, улучшения физического и психического здоровья.
Габапентин используется с 1993 г. и за первые 8 лет был назначен 5,5 млн больных СД. Такое широкое распространение препарата обусловлено его эффективностью и хорошей переносимостью. Из побочных реакций наиболее часто отмечаются головокружения, сонливость, диарея, тошнота, периферические отеки. Общая частота отрицательных эффектов не превышает 5—6% [13]. На белорусском рынке зарегистрирован габапентин (тебантин). Назначать тебантин рекомендуется с 300 мг/сут вечером, постепенно наращивая дозу до 1800 мг/сут. Титрование дозы проводится раз в 2—3 дня. Противопоказания для использования тебантина включают повышенную чувствительность к препарату или его вспомогательным компонентам. Специальные исследования по оценке влияния габапентина на плод не выполнялись, поэтому при беременности его нужно применять с осторожностью. При приеме габапентина следует соблюдать осторожность при психозах в анамнезе; препарат может нарушать работоспособность при работе с точными приборами и вождении автомобиля. При использовании препаратов, содержащих алюминий и магний, необходимо выдержать перерыв 2 часа до приема габапентина.
Кроме габапентина в эту группу входит более новый препарат — прегабалин (Lyrica), который обеспечивает равноценный обезболивающий эффект (до 50%) при использовании значительно более низких доз (150—600 мг/сут) в течение первой недели лечения. Одновременно прегабалин способствует улучшению сна и хорошо переносится [23, 24]. Стартовая доза прегабалина — по 75 мг 2 раза в день – постепенно повышается до 600 мг в сутки. После 7-дневного приема и достижения анальгетического эффекта дозу лекарственного препарата рекомендуется снизить.
Имеются данные об эффективности при ДН антиаритмических средств (лидокаина и мексилетина). Механизм действия основан на стабилизации мембран нейронов за счет блокады натриевых каналов. Лидокаин в виде медленных внутривенных инфузий (30 мин) в дозе 5 мг/кг эффективно уменьшает боль при ДН [34]. Антиноцицептивный эффект пероральной формы мексилетина в дозе 450—600 мг/сут был доказан в ряде двойных слепых плацебо-контролируемых исследований [30]. По шкале общей оценки боли улучшение оказалось незначительным, но отмечено достоверное уменьшение стреляющих, жгучих болей, покалывания и чувства жара. Побочные эффекты при лечении антиаритмическими средствами менее выражены по сравнению с антиконвульсантами.
Некоторые авторы рекомендуют использовать в комплексной терапии ДН местнораздражающие средства (финалгон, апизатрон, випросал, капсикам и др.), особенно при лечении жгучих поверхностных и колющих болей.
Альтернативой в достижении анальгетического эффекта является использование неопиоидных анальгетиков центрального действия, которые избирательно влияют на уровне чувствительных нейронов задних рогов спинного мозга (соанальгетики). Механизм действия препаратов этой группы основан на непрямом антагонизме к NMDA-рецепторам и агонизме по отношению к GABA-эргическим рецепторам при отсутствии влияния на рецепторы серотонина, допамина, опиатов, центральные мускаринергические и никотинергические, а также бензодиазепиновые рецепторы [19]. В результате происходит селективная активация нейрональных калиевых каналов и обеспечивается анальгетический эффект. При этом одновременно оказывается миорелаксирующее действие, что принципиально важно при болевых формах ДН. Представителем этой группы препаратов на белорусском рынке является флупиртин (катадолон), который обладает доказанным анальгетическим эффектом при болевых синдромах различной этиологии (радикулоневриты, вертеброгенные дорсопатии, постоперационный болевой синдром, онкологические заболевания, заболевания опорно-двигательного аппарата, в том числе остеопороз, миофасциальные синдромы и др.). Назначение флупиртина сопровождалось купированием острого болевого синдрома, нормализацией эмоционального состояния, ночного сна и оптимизацией качества жизни [40, 44, 54, 67]. Назначать катадолон следует по 100—200 мг 3—4 раза в день (суточная доза 600 мг). Ограничения в использовании препарата: беременность, детский возраст, тяжелая печеночная недостаточность, миастения, алкоголизм.
Немедикаментозные методы включают использование гимнастики для ног, массажа и различных физиотерапевтических методов (магнитотерапия, чрескожная электронейростимуляция, акупунктура и др.), но их эффективность не доказана в многоцентровых рандомизированных исследованиях. Эффективность физиотерапевтических воздействий, подтвержденная в малых группах и при коротком периоде наблюдения, позволяет рекомендовать их для включения в комплексную терапию ДН. В то же время необходимо проявлять осторожность в выборе физиотерапевтических средств лечения, так как нарушения чувствительности и вегетативные расстройства при ДН предрасполагают к образованию ожогов и язв.
Уход за стопами — обязательный компонент ведения ДН. Кроме самостоятельных мероприятий по уходу необходимы консультации в кабинете диабетической стопы, где в зависимости от выраженности местных изменений на стопах должны быть проведены манипуляции, снижающие риск образования трофических язв и, следовательно, гангрены. Для профилактики прогрессирования патологии и формирования синдрома диабетической стопы рекомендуется исключить или минимизировать воздействие внешних (тесная обувь, механические и термические воздействия) и внутренних факторов (повышенное подошвенное давление, образование участков омозолелостей, формирование деформации пальцев и стопы) [4—6].
При развитии осложнений ДН, в том числе синдрома диабетической стопы, в комплекс лечебных мероприятий включают использование препаратов различных групп, что определяется симптоматикой (местной или общей). Препаратами выбора являются те, которые способствуют восстановлению магистрального кровотока, снижению выраженности остеопороза, заживлению трофических язв и др. [21, 33, 64, 66].
Терапевтические подходы к лечению диабетической нейропатии, основанные на понимании патогенеза, находятся в центре внимания не только клиницистов, но и фармакологов. В качестве возможных групп лекарственных средств для терапии ДН рассматриваются ингибиторы протеинкиназы С; препараты, способные замедлить формирование конечного продукта гликозилирования (воздействующие посредством рецептора нуклеарного фактора NF -kB); нейротрофины (рекомбинантные ядерные факторы роста); γ-линоленовая кислота, содержащаяся в масле примулы; ингибиторы липидной пероксидации и др.[12, 39]. Перспективно использование ингибиторов протеинкиназы С (PKC), которая активируется при гипергликемии и одновременно сопровождается повышением продукции диацилглицерола, нарушением вазоактивности и утолщением базальной мембраны.
Патогенетический механизм ингибиторов РКС заключается в блокаде РКС на фоне внутриклеточной гипергликемии и повышения уровня диацилглицерола, что предотвращает нарушения экспрессии эндотелиальной синтетазы оксида азота и сосудистого эндотелиального фактора роста. Данные предварительных экспериментальных исследований по применению ингибитора изоформы PKC-β рубоксистаурина (ruboxistaurin) продемонстрировали его положительное влияние на функциональное состояние периферической нервной системы. В настоящее время завершены испытания, подтверждающие эффективность ruboxistaurin (32 и 64 мг) при диабетической ретинопатии, и завершаются три многоцентровых исследования по оценке его эффективности при ДН [8]. По первым результатам эффективность рубоксистаурина при диабетической нейропатии оказалась не слишком выраженной, однако выявленные в течение 3 лет изменения скорости прогрессии нейропатии были слабодостоверными [15].
В настоящее время ведутся исследования по оценке эффективности в эксперименте блокаторов альдостерона (кандесартан или эплеренон) и матрикса олигомерного протеина картилага (СОМР-Ang1) c выраженным позитивным эффектом.
Общим принципом лечения диабетической нейропатии должно быть назначение базовой патогенетической терапии с подключением дополнительного симптоматического лечения. До настоящего времени ДН является единственным осложнением сахарного диабета, лечение которого не определено международными стандартами [9, 25], что следует из отсутствия доказательной базы эффективности используемых подходов в многоцентровых долгосрочных рандомизированных исследованиях и определяет применение широкого спектра лекарственных средств.
Литература
1. Балаболкин М. И., Чернышова Т. Е., Трусов В. В., Гурьева И. В. // Диабетическая нейропатия: патогенез, диагностика, классификация, прогностическое значение, лечение: учеб.-метод. пособие. — М.: Экспертиза, 2003.
2. Галстян Г.Р., Анциферов М.Б. // Врач. —2000. —№ 1. —С. 23–29.
3. Гурьева И.В., Комелягина Е.Ю., Кузина И.В. Диабетическая периферическая сенсомоторная нейропатия: метод. рекомендации. — М., 2000.
4. Дедов И.И., Удовиченко О.В., Галстян Г.Р. // Диабетическая стопа. — М.: Практическая медицина, 2005. — С. 48–57.
5. Котов С. В., Калинин А. П., Рудакова И. Г. // Диабетическая нейропатия. — М.: Медицина, 2000. — С. 45, 139, 150.
6. Мохорт Т.В., Ромейко Д.И. // Диабетическая полинейропатия: метод. руководство. — Минск, 2000.
7. Овчинников Е.А., Рашид М.А., Куликов А.Ю. и др. // Качественная клиническая практика. — 2005. —№ 1. —С. 1—9.
8. Alello L. P. // Scientific Session IDF, 2006. Cape Town.
9. American Diabetes Association and American Academy of Neurology. Report and Recommendatiоn of the San Antonio Conference on Diabetic Neuropathy // Diabet. Care. — 1988. —Vol. 11. — P. 592—597.
10. Ametov A., Barinov A., Dick P. et al. // Diabet. Care. — 2001. —Vol. 23 (6).
11. Andersson G.B.J. // Lancet. — 1999. —Vol. 354. —P. 581—585.
12. Apfel S.C., Schwartz S., Adomato B.T. et al. // JAMA. — 2000. —Vol. 284. —P. 2215—2221.
13. Backonja M., Beydoun A., Edwards K.R. et al. // JAMA. — 1998. —Vol. 280. —P.1831—1836.
14. Backonja M. // Clin. Therapeutics. — 2003. —Vol. 25 (1). —P. 81—104.
15. Bastyr E.J., Cheng C., Price K.L. et al. // Diabetologia.— 2007. —Vol. 50 (Suppl.1). — S62 (0135).
16. Bush R., Wolf M., Moller J. et al. // Ann. of Nutrition and Metab. — 1991. —Vol. 35. —P. 292—296.
17. Bustamante J., Lodge J.K., Marcocci L. et al. // Free Radic. Biol. Med. —1998. —Vol. 24. —P. 1023—1039.
18. Boulton A.J.M., Rayaz Malik, Arezzo J.C.A., Sosenko J.M. // Diabet. Care. — 2004. —Vol. 27. —P. 1458—1486.
19. Carlsson K.H., Jurna I. // Eur. J. Pharmacol. —1988. —Vol. 151 (2). —P. 89—99.
20. DCCT Research Group: The effect of intensive diabetes therapy on the development and progression of neuropathy // Ann. Intern. Med. —1995. —Vol. 122. —P. 561–568.
21. Diabetic foot disorders: A Clinical Practice Guideline / R. G. Frykberg, D. G. Armstrong, J. Giurini et al. // J. of Foot &Ankle Surgery. —2000. —Vol. 39 (5). —P. 2—49.
22. Egbunike I.G., Chaffe B.G. // Pharmacotherapy. —1990. —Vol. 10. —P. 262—270.
23. Freeman R., Rosenstok J., Emir B. et al. // Diabetologia. — 2007. —Vol. 50 (Suppl.1). —S63 (0137).
24. Freynhagen R., Strojek K., Griesing T. et al. // Pain. — 2005. —Vol. 115. —P. 254—263.
25. Global Guidеline for Type 2 Diabetes: recommendations for standard, comprehensive, and minimal care (IDF Clinical Guidеlines Task Forse) // Diabet. Med.—2006. —Vol. 23. — P. 579—593.
26. Goldstein J., Yili L., Detke M.J. et al. // Pain.— 2005. —Vol.116. —P. 109—118.
27. Greb A., Bitsch R. // Intern. J. Clin. Pharm. & Therapeutics. — 1998. —Vol. 4. —P.216—221.
28. Greene D. A., Stevens M. J. / N. Hotta, D.A. Greene, D.J. Ward et al. (eds). Diabetic neuropathy. New concepts and insights. – Elsevier Science B.V., 1995. —P. 37–41.
29. Haupt E., Lederman H., Kopcke W. // Intern. J. Clin. Pharm. & Therapeutics. — 2005. —Vol. 2. —P. 71—77.
30. Jarvis В., Coukell A.J. // Drugs. — 1998. —Vol. 56. —P.691— 707.
31. Jermendy G. // Medicus Universalis. — 1995. —Vol. 15. —P. 217—220.
32. Jude E.B., Boulton A.J.M. // Diabet. Rev. — 1999. — Vol. 7. —P. 395—410.
33. Katouchkina A., Mokhort T., Romeiko D. // The diabetic foot. Тhird international symposium: Absractbook, Noordwijkerhout, The Netherlands, 5—8 May. —1999. – P. 29.
34. Kastmp J., Peterson P., Dejgard A. et al. // Pain. —1987. —Vol. 28. —P. 69—75.
35. Keen H., Payan J., Allawi J. et al. // Diabet. Care.— 1993. —Vol. 16. —P. 8—15.
36. Kempler P. Neuropathies. Pathomechanism, clinical presentation, diagnosis, therapy / ed. by P. Kempler. — Springer, 2002.
37. Kowlulu R.A., Odenbach S. // Diabetes. — 2004. —Vol. 53. —P. 3233—3238.
38. Lin J., Bierhaus A., Bugert P. et al. // Diabetologia. — 2006. —Vol.49. —P.1089—1096.
39. Litchy W., Dyck P.J., Tesfaye S., Zhang D. // Diabetes. —2002. —Vol. 45 (Suppl. 2). —S197.
40. Luben V., Muller H., Lobisch M. et al. // Fortschr. Med. —1994. —Vol.112 (19). —P. 31—38.
41. Mayhew J.A., Gillon K.R., Hawthorne J.N. // Diabetologia. —1983. —Vol. 24. —P. 13—15.
42. Мах M. В., Culnane M., Schafer S.C. et al. // Neurology. — 1987. —Vol. 37. —P. 589—596.
43. Mendell J.R., Sahenk Z. // New Engl. J. Med. — 2003. —Vol. 248. —P. 1243–1255.
44. Mueller-Schwefe G. // Fortschr. Med. Origin. — 2003. —Vol. 121 (1). —P. 11—18.
45. New J.P., Hollis S., Campbell F. et al. // Diabetologia. — 2000. —Vol. 43. —P. 836—843.
46. Oates P.J., Klioze S.S. & Zopolrestzat Diabetic Neuropathy Study Group // Diabetologia. — 2007. —Vol. 50 (Suppl.1). — S62 (0136).
47. Obrosova I.G., Minchenko A.G., Marinescu V. et al. // Diabetologia. — 2001. —Vol. 44. —P.1102—1110.
48. Packer L. // Antioxidants in Diabetes Management / eds. L. Packer et al. — New York, 2000. — P. 1–15.
49. Pfeifer M.A., Schumer M.P., Gerber D.A. // Diabetes. — 1995. —Vol. 44. —P. 1355—1361.
50. Raskin J., Princhett Y.L., Wang F. et al. // Pain Medicine. — 2005. —Vol. 6 (5). —P. 346—356.
51. Raskin J., Smith T.R., Wong K. et al. // J. of Palliative Med. — 2006. —Vol. 9 (1). —P. 29—39.
52. Reichel G. Diabetische Neuropathie: Bedeutung von Hyperglykamia und oxydativem Stress. Joint Spring Meeting. Abstract. —Acta Medic AG. Munchen, 17–19 Marz, 1995.
53. Reljanovic M., Reichel G., Rett K. et al. // Free Rad. Res. — 1998. —Vol. 7. —P. 51—57.
54. Ringer J., Miethe D., Pittrow D. et al. // Pain Fortschr. Med. — 2003. —Vol. 53(7). —P. 496—502.
55. Ryan C.M. // Diabetologia. — 2006. —Vol. 49. —P. 2229—2233.
56. Said G. Diabetic Neuropathy / A.J.M. Boulton, ed. — Cologne, Aventis, Academy Press, 2001. —P. 16—41.
57. Skyler J. // Endocrinol. Metab. Clin. — 1996. —Vol. 25. —P. 243—254.
58. Stracke H., Lindermann A., Federlin K. // Endocrinol. and Diabetes. —1996. —Vol.104. — P. 311—316.
59. Tanenberg R., Schummer M., Green D., Pfeifer M. // The Diabetic Foot, 6th ed. — Mosby, 2001. — P. 33—64.
60. The Diabetes Control and Complications Trial Research Group. The absence of a glycemic threshold for the development of long-term complications: the perspective of the Diabetes Control and Complications Trial // Diabetes. — 1996. —Vol. 45. —P. 1289—1298.
61. Thomas P.K. Classification of the diabetic neuropathies. Textbook of Diabetic Neuropathy / F. A. E. Gries, P.A. Low, D.Ziegler (eds.). — Stuttgart, Thieme, 2003. —P. 175–177.
62. UK Prospective Diabetes Study (UKPDS 34) // Lancet. —1998. —Vol. 352. —P.854—865.
63. Vinik A.I., Park T.S., Stansberry K.B., Pittenger G.L. // Diabetologia. — 2000. —Vol.43. —P. 957—973.
64. Ward J.D. // Diabet. Care. — 1999. —Vol. 22 (2). —P. B84—B88.
65. Watkins P. J., Thomas P. K. // J. Neurol. Neurosurg. Psychiatr. —1998. —Vol. 65. —P. 620—633.
66. Williams G., Pickup C. Handbook of diabetes. — London, 1996.
67. Worz R., Lobish M., Schwiman B. et al. // Fortschr. Med. —1995. —Vol.113 (32). —P.47—52.
68. Ziegler D., Hanefeld M., Ruhnau K.J. et al. // Diabetologia. — 1995. —Vol. 38. —P. 1425—1433.
69. Ziegler D., Hanefeld M., Ruhnau K.J. et al. // Diabet. Care.—1999. —Vol. 8. — P. 1296—1301.
70. Ziegler D., Low P.A., Boulton A.J.M. et al. // Diabetologia. — 2007. —Vol. 50 (Suppl.1). —P. S63 (0138).
71. Ziegler D., Scharz H., Conrad F. et al. // Diabet. Care. —1997. —Vol.20. —P. 369—373.
72. Ziegler D., Reljianovic M., Mehnert H., Gnes F. A. // Exp. Clin. Endocrinol. Diabetes. — 1999. —Vol. 107. —P. 421—430.
73. Ziegler D., Nowak H., Kempler P. et al. // Diabet. Med. — 2004. —Vol. 21. — P. 114—121.
Медицинские новости. – 2008. – №1. – С. 40-47.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.