.jpg)
Trisvetova E.L.
Belarusian State Medical University, Minsk
Causes, risk factors and medication prevention of chronic heart failure
heritabledisordersofconnectivetissue
Резюме. Сердечно-сосудистые изменения считают ведущей патологией, часто определяющей качество и прогноз жизни при наследственных нарушениях соединительной ткани. Исходом аномалий клапанного аппарата сердца, аневризмы/диссекции аорты при синдроме Марфана, митральной регургитации при пролапсе митрального клапана с миксоматозом является хроническая сердечная недостаточность, к которой приводит и выявленное недавно поражение миокарда – первичная кардиомиопатия. Медикаментозное лечение бета-блокаторами, ингибиторами АПФ и с доказанной эффективностью блокатором рецепторов ангиотензина II – лозартаном применяют для замедления прогрессирования расширения аорты, миксоматоза и улучшения функции миокарда.
Ключевые слова: наследственные нарушения соединительной ткани, хроническая сердечная недостаточность, кардиомиопатия, медикаментозное лечение, лозартан.
Медицинские новости. – 2016. – №3. – С. 23–28.
Summary. Cardiovascular changes considered a leading pathology, often determines the quality of life and prognosis of hereditary disorders of connective tissue. Exodus valvular abnormalities, aneurysm/dissection of the aorta in Marfan syndrome, mitral regurgitation with mitral valve prolapse with myxomatosis is a chronic heart failure, which leads to the recently revealed lesion infarction – primary cardiomyopathy. Drug therapy with beta-blockers, ACE inhibitors, and with proven efficacy, an angiotensin II receptor blocker – losartan, used to slow the progression of aortic enlargement, myxomatosis and improved myocardial function.
Keywords: hereditary connective tissue disorders, chronic heart failure, cardiomyopathy, medication, losartan.
Meditsinskie novosti. – 2016. – N3. – P. 23–28.
Исходом многих сердечно-сосудистых заболеваний является хроническая сердечная недостаточность (ХСН), ассоциированная с высокой заболеваемостью, прогрессирующим течением и смертностью [1]. По результатам исследований, выполненных в США и Европе в разных когортах населения, сердечную недостаточность регистрируют в 1–12% случаев [1–3]. Известно, что распространенность сердечной недостаточности увеличивается с возрастом и встречается чаще у мужчин, чем у женщин (рис. 1) [1, 4]. После установления диагноза сердечной недостаточности с дисфункцией левого желудочка выживаемость пациентов через 5 и 10 лет составляет 50 и 10% соответственно [5, 6].
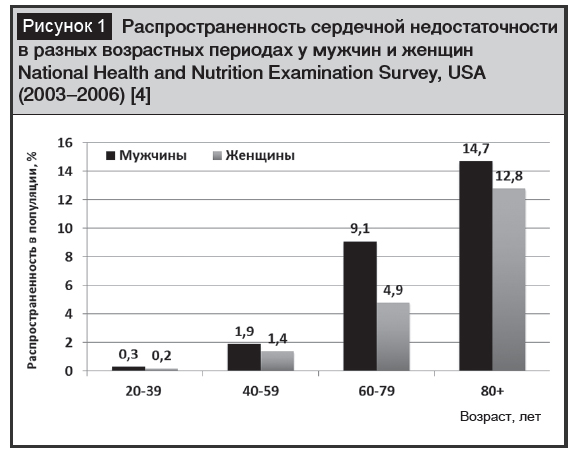
Появление сердечной недостаточности повышает риск смертности в 4 раза, а госпитальная летальность находится на уровне 7–10% [4]. Рост распространенности сердечной недостаточности ожидается в дальнейшем в связи со старением населения, продлением жизни кардиологических пациентов, терапевтическими и хирургическими методами лечения и возрастанием в популяции основных факторов риска развития ХСН – ишемической болезни сердца (ИБС), артериальной гипертензии (АГ), ожирения и сахарного диабета [7, 8].
По результатам исследования Framingham, основные этиологические причины развития ХСН – сердечно-сосудистые заболевания, включающие АГ, ИБС, сочетание АГ и ИБС, ревматические болезни сердца (РБ), сочетание РБ и АГ, а также смешанные факторы (рис. 2) [9].
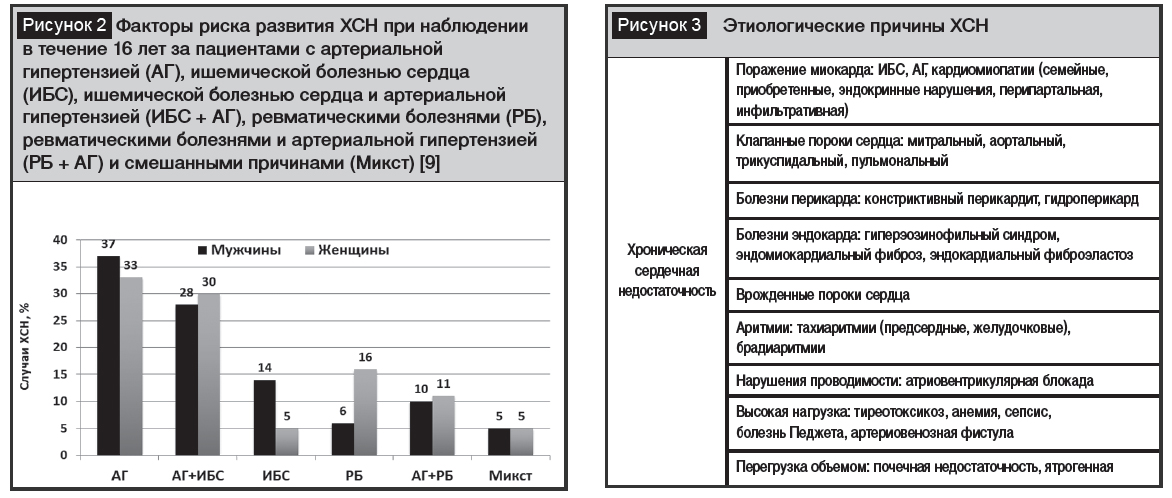
В XXIвеке изменилась структура и соотношение факторов риска развития ХСН [10]. Исследования, выполненные в Российской Федерации, свидетельствуют о том, что основными причинами госпитализации пациентов с ХСН являются АГ – 88%, ИБС – 59% [11, 12]. Пороки сердца как причина развития ХСН встречаются реже – в 4,3%, миокардиты – в 3,6%, дилатационная кардиомиопатия – в 0,8% случаев. В Национальных рекомендациях ОССН, РКО и РНМОТ по диагностике и лечению ХСН, помимо основных причин, отмечено утяжеление течения ХСН постоянной формой фибрилляции предсердий в 10,3% случаев [11, 12].
Известны многие другие причины развития ХСН, часто встречающиеся как комбинации нескольких факторов (рис. 3).
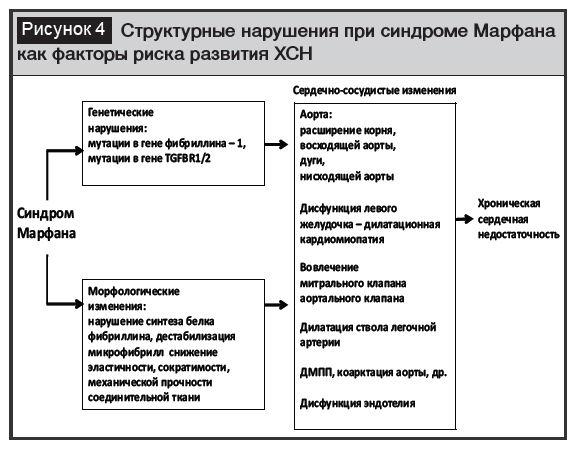
Определенный вклад в заболеваемость ХСН вносят генетические болезни. С конца XXвека активно изучаются генетические аспекты, особенности клинических проявлений, причины и факторы риска развития осложнений и ассоциированных состояний, патогенетические и симптоматические методы лечения при наследственных нарушениях соединительной ткани (ННСТ) [13–15].
ННСТ представлены синдромами и фенотипами, клиническая диагностика которых основывается на выявлении совокупности характерных признаков, обусловленных изменениями строения и биомеханических свойств соединительной ткани [16, 17]. Системные признаки ННСТ проявляются изменениями в двух и более системах организма, в том числе аномалиями сердечно-сосудистой системы, вызывающими развитие ХСН, нередко определяющими качество и прогноз жизни.
Классическим представителем ННСТ, наиболее распространенным (1:3000 – 5000 населения) и изученным является синдром Марфана – генетическое заболевание, при котором, несмотря на поражение многих систем организма, именно сердечно-сосудистые изменения в случае отсутствия комплексного (немедикаментозного, медикаментозного, хирургического) лечения в конечном итоге приводят к дилатации сердца и сердечной недостаточности с высокой смертностью вследствие ее прогрессирования, внезапной сердечной смерти или злокачественной аритмии [18, 19].
Существующие современные представления о структурных макро- и микроскопических, биохимических и молекулярно-генетических особенностях при синдроме Марфана лежат в основе обобщенного представления о факторах, приводящих к появлению ХСН [20].
Нарушения функции сердечно-сосудистой системы, как и других систем организма, обусловлены изменениями свойств соединительной ткани, возникающими в результате мутаций в гене FBN1, расположенном в хромосоме 15q21.1. Фибриллин, гликопротеин, – один из основных строительных белков микрофибрилл, формирующих коллаген внеклеточного матрикса соединительной ткани. Видоизмененный FBN1 нарушает структуру, снижает транспорт, уменьшает количество микрофибрилл, обеспечивающих упругие свойства соединительной ткани [18, 20].
Известно, что мутации в гене FBN1 вызывают снижение механических свойств соединительной ткани. В ходе экспериментальных исследований H.C. Dietz и соавт. показали, что многие легочные, сердечно-сосудистые, скелетные и мышечные аномалии при синдроме Марфана возникают вследствие нарушения активации сигнального пути, регулируемого многофункциональным цитокином – трансформирующим фактором роста? (TGF -?)[21]. TGF -??– регулятор морфогенеза, пролиферации, апоптоза, формирования внеклеточного матрикса, активации матриксных металлопротеиназ 2 и 9. Нарушения в регуляции TGF -??у пациентов с синдромом Марфана относятся к одному из механизмов развития миксоматоза клапанов сердца, повышения уровня апоптоза в легочной ткани с образованием кист и спонтанного пневмоторакса, а также расширения корня аорты. Установлено, что в семьях или у спорадических пациентов с синдромом Марфана без эктопии хрусталика определяются мутации в одном из двух генов (TGF -? R1 и TGF -? R2), которые кодируют рецепторы цитокина TGF -? [20].
Сердечно-сосудистые изменения, в первую очередь описываемые при синдроме Марфана, включают аневризму/диссекцию аорты, дилатацию ствола легочной артерии, пролабирование створок митрального и/или трикуспидального клапанов, кальцификацию митрального кольца [21, 22]. Исследователи отмечают другие, редко встречающиеся сердечно-сосудистые проявления: коарктацию аорты, дефект межпредсердной перегородки, открытый артериальный проток, патологическую извитость артерий [18]. Нарушения внутрисердечной гемодинамики в результате прогрессирующей митральной и/или аортальной недостаточности приводят к развитию ХСН.
В последние годы при синдроме Марфана в тех случаях, когда отсутствует тяжелая клапанная патология, описывают кардиомиопатию [22–24]. Доказательства наличия дилатационной кардиомиопатии получены F. Alpendurada и соавт. в результате комплексной оценки функции сердца с применением метода магнитно-резонансной томографии (МРТ) крупнейшей когорты пациентов (n=68) с синдромом Марфана без значительной клапанной регургитации и кардиохиругического лечения [22]. У 25% пациентов в возрасте 33,9±11,9 года (18–71) исследователи отметили увеличение конечного диастолического и систолического объема, уменьшение фракции выброса левого желудочка (53,8±3,1 против 65,2±4,5%), а также увеличение конечного систолического и диастолического объема правого желудочка. В результате исследования были сделаны выводы о наличии кардио-миопатии с нарушением систолической функции левого и правого желудочка, протекающей бессимптомно и не зависящей от классических сердечно-сосудистых проявлений и лекарственного сопровождения (преимущественно ?-блокаторы) во время исследования. Вместе с тем уровень фракции выброса у пациентов, получавших медикаментозную терапию, был выше по сравнению с результатами не лечившихся (63,7±6,9 против 59,7±6,4%; р=0,02).
В исследованиях с применением эхокардиографического метода с тканевой допплерографией также наблюдали изменения размеров и незначительное ухудшение систолической и диастолической функции левого желудочка у пациентов с синдромом Марфана [18, 23]. Вполне закономерно, что при МРТ были получены доказательства увеличения конечного систолического объема (с поправкой на площадь поверхности тела) и снижения фракции выброса левого желудочка. МРТ идентифицирует пролапса митрального клапана (ПМК) с 100% чувствительностью и специ-фичностью, обеспечивая количественное определение объемов и функции желудочков [24].
Дилатация левого желудочка при синдроме Марфана, помимо развития ХСН, ассоциируется с нарушениями реполяризации и злокачественными желудочковыми аритмиями, ухудшающими функцию миокарда [18, 20].
Скелетные аномалии также вносят свой вклад в появление признаков ХСН. Недавние исследования методом МРТ показали, что Pectus excavatum (при высокой степени экскавации) приводит к снижению диастолической функции левого желудочка, улучшение которой отмечают после хирургической коррекции деформации грудной клетки [25].
По мнению H.T. Syyong и соавт., для синдрома Марфана характерна эндотелиальная дисфункция, обусловленная изменениями структуры сосудистой стенки в результате дегенерации эластиновых волокон, обеспечивающих растяжимость и эластичность [26]. Результаты экспериментальных исследований показали, что при прогрессировании синдрома Марфана отмечается повышение жесткости и нарушение вазомоторной функции эндотелия со снижением вазорелаксации.
Таким образом, прогрессирующее течение синдрома Марфана и сердечно-сосудистых изменений приводит к появлению признаков бивентрикулярного поражения сердца с нарушениями диастолической и систолической функции (рис. 4).
Менее выраженные по сравнению с синдромом Марфана проявления нарушения строения и метаболизма соединительной ткани отмечают при синдроме ПМК. Первичное пролабирование створок митрального клапана (несиндромный ПМК, то есть не относящийся к проявлениям синдрома Марфана, Элерса – Данло, Loeys – Dietz, несовершенного остеогенеза, эластической псевдоксантомы, аневризмы – остеоартрита и гипертрофической кардиомиопатии) относится к генетическим синдромам ННСТ, встречается как спорадический или семейный тип с аутосомно-доминантным типом наследования и вариабельностью пенетрантности 30–50%, а также Х-сцепленное. Известны три локуса на хромосомах 16p12.1-р11.2, 11p15.4 и 13q31.3-р32.1, сцепленных с ПМК, но специфический ген не описан. По меньшей мере, 16 генов известны в регионе, которые могут быть ответственны за клапанное ремоделирование [27].
Влияние на строение внеклеточного матрикса и развитие миксоматоза (генетически обусловленное разрушение нормальной архитектоники фибриллярных, коллагеновых и эластических структур соединительной ткани) оказывает избыточный коллаген III типа, обусловливающий снижение уровня молекулярной организации коллагеновых волокон (рис. 5). Активация сигнальных путей TGF -? вызывает дисрегуляцию компонентов внеклеточного матрикса: интерстициальные клетки приобретают свойства активированных миофибробластов, характеризующихся экспрессией виментина и альфа-актина гладких мышц, способствующих повышению концентрации протеолитических ферментов (металлопротеиназ), которые разрушают коллаген и эластин со скоростью, превышающей их синтез [28, 29].

По результатам эхокардиографического исследования ПМК в популяции встречается с частотой 1–2,5%, по результатам исследования Framingham – 2,4%, при аутопсии – 8% [27, 30].
Первичный ПМК – синдром, проявлением которого является смещение митральной створки (-ок) более 2 мм (не менее 3 мм, по данным российских и белорусских исследователей) выше уровня фиброзного кольца митрального клапана в левое предсердие во время систолы (по результатам эхокардиографического исследования) в сочетании с клиническими признаками системного вовлечения соединительной ткани [16, 27].
Длительное время ПМК может протекать бессимптомно или малосимптомно, вместе с тем осложнения, возникающие в молодом возрасте, нередко влияют на прогноз жизни и включают инфекционный эндокардит, нарушения ритма и проводимости сердца, внезапную сердечную смерть, тромбоэмболию сосудов головного мозга (рис. 6). Прогрессирование ПМК с признаками миксоматоза, порхающей створкой митрального клапана и/или митральной регургитацией сопровождается увеличением левого желудочка и предсердия, развитием ХСН часто у пациентов среднего возраста.
В патогенезе ПМК и его осложнений важное место отводят миксоматозу. В хирургической литературе рассматривают два основных гистологических фенотипа миксоматоза: болезнь Барлоу и фиброэластический дефицит. При болезни Барлоу ХСН развивается в молодом и среднем возрасте пациентов вследствие митральной недостаточности из-за деформации, потери эластичности и значительного выбухания створок митрального клапана, расширения митрального кольца и удлинения створочных хорд с редкими их разрывами. При фиброэластическом дефиците пациенты с митральной недостаточностью и ХСН в более старшем возрасте нуждаются в хирургическом лечении, створки митрального клапана при макроскопическом исследовании тонкие и гладкие, умеренная дилатация митрального кольца и часто разрывы створочных хорд с задней порхающей створкой митрального клапана. До настоящего времени не выяснено, относятся данные два гистологических фенотипа к одному синдрому или к двум генетически различным заболеваниям [32, 33].
Результаты клинических исследований последних лет показали, что у молодых людей с ПМК без гемодинамически значимой митральной регургитации и порхающей створки митрального клапана также наблюдают нарушения систолической и/или диастолической функции миокарда, свидетельствующие о наличии кардиомиопатии [31, 32, 34, 35].
Исследование функционального состояния миокарда 78 бессимптомных пациентов (женщины – 28%, мужчины – 72%) 19,7±1,6 года при первичном ПМК показало, что в 25% случаев у пациентов с миксоматозом митрального клапана и минимальной митральной регургитацией наблюдают дилатацию левого желудочка, ухудшение систолической и диастолической функции [35]. При применении методики speckle tracking выявлено снижение всех составляющих деформации: скорости деформации и ротации миокарда межжелудочковой перегородки, передней стенки, верхушки; повышение показателей в области боковой, задней и нижней стенки левого желудочка, обусловленное аномальной тракцией папиллярных мышц [35, 36]. В случае применения медикаментозной терапии, включающей ингибиторы АПФ и блокаторы рецепторов ангиотензина II, влияющих на активность TGF -???отмечено замедление прогрессирования миксоматоза.
За последнее десятилетие неоднократно пересматривали систематизацию «недифференцированных», «несиндромных» или многофакторных ННСТ, не являющихся моногенными синдромами [16, 17]. Несмотря на различную терминологию, исследователи формировали группы, ориентируясь на малые аномалии сердца, количество признаков дисморфогенеза более 6, дисморфогенетические изменения в двух и более системах, в неоднородных группах пациентов выявлены структурные изменения левого желудочка, гемодинамические особенности и дисфункция эндотелия [37–41].
В группе практически здоровых молодых мужчин с малыми аномалиями сердца (ПМК без митральной регургитации и миксоматоза, аномально расположенные хорды, аномалии папиллярных мышц и др.) и дисморфогенетическими внешними и висцеральными признаками при ультразвуковом исследовании сердца выявлено увеличение размеров левого предсердия и тенденция к нарушению диастолической функции миокарда левого желудочка, на которую влиял топографический вариант аномально расположенной хорды, изменяющий геометрию левого желудочка [37].
Дисфункция эндотелия при выполнении пробы с реактивной гиперемией у молодых мужчин с многофакторными ННСТ без манифестирующих клинически заболеваний внутренних органов характеризовалась нарушением эндотелий-зависимой и неизмененной эндотелий-независимой функции [40].
Несомненным доказательством развития дилатационной кардиомиопатии при многофакторных ННСТ (дисплазии соединительной ткани) с митральной или трикуспидальной регургитацией являются наблюдения С.Л. Дземешкевича и соавт. [42]. При оперативном лечении пациентов с дилатационной кардиомиопатией и ХСН в 25% случаев макроскопически и при биопсии миокарда и створок клапанов сердца (митрального и трикуспидального) исследователи наблюдали признаки, свидетельствующие о дисплазии соединительной ткани. Авторы пришли к выводу о единой природе механизмов поражения клапанов сердца и миокарда при дисплазии соединительной ткани.
Таким образом, факторы риска развития и признаки ХСН выявляют не только при моногенных, но и многофакторных фенотипах ННСТ, о признаках которых следует помнить практическому врачу. Результаты экспериментальных и клинических исследований доказывают структурные и функциональные нарушения в клапанном аппарате сердца и внеклеточном матриксе миокарда при синдроме ПМК, функциональные нарушения, дисфункцию эндотелия – при многофакторных фенотипах.
Вопрос о медикаментозной профилактике и лечении ХСН при моногенных синдромах и многофакторных фенотипах ННСТ для практической медицины сложный. В лечении сердечно-сосудистых осложнений при синдроме Марфана применяют препараты, замедляющие прогрессирование расширения аорты – ?-адреноблокаторы, ингибиторы АПФ и блокаторы рецепторов ангиотензина II.
Начиная с 2006 года, приоритетным направлением в лечении расширения аорты при синдроме Марфана считают применение блокатора рецепторов ангиотензина II – лозартана в дозе 100 мг/сутки, показавшему замедление прогрессирования расширения торакальной аорты на уровне корня и сино-тубулярного соединения в результате подавления активности сигнальных путей TGF -? [43]. Тот же механизм действия лозартана является перспективным в отношении замедления прогрессирования миксоматоза.
Вместе с тем не решены вопросы о показаниях к назначению препарата при пограничной ширине корня аорты при синдроме Марфана, возможности эффективного лечения при синдромах, сопровождающихся аневризмой/диссекцией аорты; не известна степень миксоматоза, диагностированная по ультразвуковым признакам, при которой следует начинать терапию лозартаном, и заболевания, при которых возможно эффективное лечение; отсутствуют наблюдения по продолжительности лечения, возможной комбинации с другими лекарственными средствами, влияющими на иные патогенетические механизмы ННСТ, улучшающими структуру соединительной ткани.
Л И Т Е Р А Т У Р А
1. John, J.V. Рекомендации Европейского общества кардиологов (ЕОК) по диагностике и лечению острой и хронической сердечной недостаточности 2012 / J.V. John, V. McMurray, S. Adamopoulos, S.D. Anker [et al.] // Российский кардиологический журнал. – 2012. – Vol.4 (102), приложение 3. – P.1–68.
2. Mosterd, A. Clinical epidemiology of heart failure / A. Mosterd, A.W. Hoes // Heart. – 2007. – Vol.93. – P.1137–1146.
3. Roger, V. Epidemiology of Heart Failure / V. Roger // Circulation Research. – 2013. – Vol.113. – P.646–659.
4. Lloyd-Jones, D. Heart disease and stroke statistics-2010 update: a report from the American Heart Association / D. Lloyd-Jones [et al.] // Circulation. – 2010 – Vol.121. – P.46–215.
5. MacIntyre, K. Evidence of improving prognosis in heart failure: trends in case fatality in 66 547 patients hospitalized between 1986 and 1995 / K. MacIntyre, S. Capewell, S. Stewart [et al.] // Circulation. – 2000. – Vol.102. – P.1126–1131.
6. Mosterd, A. The prognosis of heart failure in the general population: The Rotterdam Study / A. Mosterd, B. Cost, A.W. Hoes [et al.] // Eur. Heart. J. – 2001. – Vol.22. – P.1318–1327.
7. Yach, D. The global burden of chronic diseases: overcoming impediments to prevention and control / D. Yach, C. Hawkes, C.L. Gould [et al.] // JAMA. – 2004. – Vol.291. – P.2616–2622.
8. Yusuf, S. Global burden of cardiovascular diseases: Part II: variations in cardiovascular disease by specific ethnic groups and geographic regions and prevention strategies / S. Yusuf, S. Reddy, S. Ounpuu [et al.] // Circulation. – 2001. – Vol.104. – P.2855–2864.
9. McKee, P.A. The natural history of congestive heart failure: the Framingham study / P.A. McKee, W.P. Castelli, McNamara [et al.] // N. Engl. J. Med. – 1971. – Vol.285. – P.1441–1446.
10. Ponikowski, P. Heart failure: preventing disease and death worldwide. ESC / P. Ponikowski, S.D. Anker, K.F. AlHabib [et al.] // Heart. Failure. – 2014. – Vol.1 (1). – P.4–25.
11. Фомин, И.В. Эпидемиология хронической сердечной недостаточности в Российской Федерации. В кн.: Хроническая сердечная недостаточность / И.В. Фомин, Ф.Т. Агеев [и др.]. – М., 2010. – С.7–77.
12. Мареев, В.Ю. Национальные рекомендации ОССН, РКО и РНМОТ по диагностике и лечению ХСН (четвертый пересмотр) / В.Ю. Мареев, Ф.Т. Агеев, Г.П. Арутюнов [и др.] // Журнал сердечной недостаточности. – 2013. – №7 (14, 81). – С.379–472.
13. Devereux, R.B. Recent developments in the diagnosis and management of mitral valve prolapsed / R.B. Devereux // Curr. Opin. Cardiol. – 1995. – Vol.10. – P.107–116.
14. Bouknight, D.P. Current management of mitral valve prolapsed / D.P. Bouknight, R.A. O’Rourke // Am. Fam. Physician. – 2000. – Vol.61. – P.3343–3350, 3353–3354.
15. Milewicz, D.M. Genetic disorders of the elastic fiber system / D.M. Milewicz, Z. Urban, C. Boyd // Matrix. Biol. – 2000. – Vol.19. – P.471–480.
16. Наследственные и многофакторные нарушения соединительной ткани. Национальные рекомендации. – Минск, 2015. – 48 с.
17. Кадурина, Т.И. Дисплазия соединительной ткани. Руководство для врачей / Т.И. Кадурина, В.Н. Горбунова. – СПб., 2009. – 714 с.
18. De Backer, J. Cardiovascular characteristics in Marfan syndrome and their relation to the genotype / J. De Backer // Verh. K. Acad Geneeskd Belg. – 2009. – Vol.71 (6). – P.335–371.
19. Dietz, H.C. Marfan Syndrome // H.C. Dietz, R.A. Pagon, M.P. Adam, H.H. Ardinger, S.E. Wallace, A. Amemiya, L.J.H. Bean, T.D. Bird, C.T. Fong, H.C. Mefford, R.J.H. Smith, K. Stephens (eds) // Source Gene Reviews® [Internet]. Seattle (WA): University of Washington, Seattle, 1993–2016. – 2001. – Apr. 18 [updated 2014, Jun. 12].
20. Loeys, B. The revised Ghent nosology for the Marfan syndrome / B. Loeys, H.C. Dietz, A.C. Braverman [et al.] // J. Med. Genet. – 2010. – Vol.47. – P.476–485.
21. Alpendurada, F. Evidence for Marfan cardiomyopathy / F. Alpendurada, J. Wong, A. Kiotsekoglou [et al.] // Eur. J. of Heart Failure. – 2010. – Vol.10 (12). – P.1085–1091.
22. Kiotsekoglou, A. Impaired right ventricular systolic function demonstrated by reduced atrioventricular plane displacement in adults with Marfan syndrome / A. Kiotsekoglou, G.R. Sutherland, J.C. Moggridge [et al.] // Eur. J. Echocardiogr. – 2009. – Vol.10 (2). – P.295–302.
23. Cook, J.R. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome / J.R. Cook, L. Carta, L. Bеnard [et al.] // J. Clin. Invest. – 2014. – Vol.124, N3. – P.1329–1339.
24. Zagrosek, AVK-B.F. Hemodynamic impact of surgical correction of pectusexcavatum – a cardiovascular magnetic resonance study / AVK-B.F. Zagrosek, S. Polleichtner, J. Schulz-Menger // J. Cardiovasc. Magn. Reson. – 2011. – Vol.13 (Suppl. 1). – P.190.
25. Dormand, H. Cardiovascular Magnetic Resonance in Marfan syndrome / H. Dormand, R. Mohiaddin // J. Cardiovasc. Magnetic resonance. – 2013. – Vol.15. – P.33. – doi: 10.1186/1532-429X-15-33.
26. Syyong, H.T. Dysfunction of endothelial and smooth muscle cells in small arteries of a mouse model of Marfan syndrome / H.T. Syyong, A.W.Y. Chung, H.H.C. Yang [et al.] // British Journal of Pharmacology. – 2009. – Vol.6 (158). – P.1597–1608.
27. Bonov, R.O. ACC/AHA Guidelines for the management of patients with valvular heart disease / R.O. Bonov, B. Carabello, A.C.Jr. de Leon [et al.] // JACC. – 1998. – Vol.5 (32). – P.1486–1588.
28. Rabin, E. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves / E. Rabin, M. Aikawa, J.R. Stone [et al.] // Circulation. – 2001. – Vol.104. – P.2525–2532.
29. Bischoff, J. Progenitor cells confer plasticity to cardiac valve endothelium / J. Bischoff, E. Aikawa // J. Cardiovasc. Transl. Res. – 2011. – Vol.4. – P.710–719.
30. Трисветова, Е.Л. Анатомия малых аномалий сердца / Е.Л. Трисветова, О.А. Юдина. – Минск, 2006. – 96 с.
31. Boudoulas, K.D. Floppy Mitral Valve (FMV)/Mitral Valve Prolapse (MVP) and the FMV/MVP Syndrome: Pathophysiologic Mechanisms and Pathogenesis of Symptoms / K.D. Boudoulas, H. Boudoulas // Cardiology. – 2013. – Vol.126. – P.69–80.
32. Rizzo, S. TGF-beta1 pathway activation and adherens junction molecular pattern in nonsyndromic mitral valve prolapsed / S. Rizzo, C. Basso, E. Lazzarini // Cardiovasc. Pathol. – 2015. – Vol.24 (6). – P.359–367.
33. Delling, F.N. Epidemiology and Pathophysiology of Mitral Valve Prolapse / F.N. Delling, R.S. Vasan // Circulation. – 2014. – Vol.129. – P.2158–2170.
34. Attenhofer, J.C. Ventricular cardiomyopatihy in mitral valve prolapse: factor or fiction? / J.C. Attenhofer, M. Greutmann, H.M. Connolly [et al.] // EMJ Cardiol. – 2015. – Vol.3 (1). – P.30–37.
35. Malev, E. Evaluation of left ventricular systolic function in young adults with mitral valve prolapsed / E. Malev, E. Zemtsovsky, А. Pshepiy [et al.] // Experimental and clinical cardiology. – 2012. – Vol.4 (17). – P.165–168.
36. Малев, Э.Г. Ремоделирование миокарда и диастолическая дисфункция левого желудочка при пролапсе митрального клапана / Э.Г. Малев, А.Р. Пшепий, Л.В. Васина [и др.] // Российский кардиологический журнал. – 2013. – №2 (100). – С.12–17.
37. Трисветова, Е.Л. Малые аномалии сердца (клиника, диагностика, экспертное значение у мужчин молодого возраста) / Е.Л. Трисветова. – Минск, 2005. – 200 с.
38. Нечаева, Г.И. Дисплазия соединительной ткани: терминология, диагностика, тактика ведения пациентов / Г.И. Нечаева, И.А. Викторова. – Омск, 2007. – 188 с.
39. Трисветова, Е.Л. Особенности сосудодвигательной функции эндотелия у мужчин молодого возраста / Е.Л. Трисветова, Н.М. Вараницкая, Р.Ф. Ермолкевич // Дисфункция эндотелия: экспериментальные и клинические исследования. Труды V Международной научно-практ. конф. 22–23 мая 2008 г. – Витебск: ВГМУ, 2008. – С.217–220.
40. Трисветова, Е.Л. Особенности эндотелиальной функции по результатам пробы с реактивной гиперемией у мужчин призывного возраста с наследственными нарушениями соединительной ткани / Е.Л. Трисветова, О.А. Паторская, Н.В. Томчик; отв. ред. В.В. Зинчук // Вопросы экспериментальнои? и клиническои? физиологии: сб. науч. трудов, посвященныи? 100-летию со дня рождения Аринчина Николая Ивановича. – Гродно, 2014. – С.301–304.
41. Нечаева, Г.И. Торакодиафрагмальное сердце при дисплазиях соединительнои? ткани – природно-экспериментальная модель диастолическои? дисфункции / Г.И. Нечаева, И.А. Викторова // Серднедост. – 2001. – №6 (1). – С.27–33.
42. Дземешкевич, С.Л. Дисплазия митрального клапана как компонент синдрома дилатационной кардиомиопатии / С.Л. Дземешкевич, Ю.В. Фролова, Д.Н. Федоров [и др.]. – Клин. и эксперимент. хир. журн. им. акад. Б.В. Петровского. – 2015. – №2.– С.18–24.
43. Groenink, M. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: A randomized controlled trial / M. Groenink, A.W. den Hartog, R. Franken [et al.] // Eur. Heart. J. – 2013. – Vol.34. – P.3491–3500.
Медицинские новости. – 2016. – №3. – С. 23-28.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.