Shevchenko А.А.
Institute of pediatrics, obstetrics and gynecology of the National academy of medical sciences of Ukraine, Kyiv
Hemimegalencephaly in the structure of the congenital malformations
of the central nervous system
Резюме. Приведены основные сведения о редкой и тяжелой врожденной мальформации ЦНС – гемимегалэнцефалии, рассматриваются ее основные клинические и диагностичекие аспекты. Обоснована необходимость применения магнитно-резонансной томографии с целью оптимизации пренатальной диагностики врожденной патологии ЦНС и акушерской тактики для уменьшения детской смертности и первичной инвалидности. Представлено описание клинического случая: пациент с данной патологией проходил обследование и лечение в клинике детской психоневрологии Института педиатрии, акушерства и гинекологии НАМН Украины.
Ключевые слова: гемимегалэнцефалия, врожденные мальформации ЦНС, диагностика, лечение, дети.
Медицинские новости. – 2014. – №6. – С. 61–65.
Summary. Presents basic information about rare and severe congenital malformation of the central nervous system (CNS)-hemimegalencephaly with a focus on the basic clinical, diagnostic and curative aspects. It was grounded the necessity of the use of magnetic resonance imaging for optimization of the prenatal diagnostics of congenital pathology of the CNS and obstetric tactics in order to reduce child mortality and primary disability. It was described clinical case of patient with this pathology during examination and treatment at the department of childrens’ psychoneurology of the Institute of pediatrics, obstetrics and gynecology of the National academy of medical sciences of Ukraine.
Keywords: hemimegalencephaly, congenital malformation of the CNS, diagnostics, treatment, children.
Meditsinskie novosti. – 2014. – N6. – P. 61–65.
Врожденные мальформации центральной нервной системы (ЦНС) являются актуальной проблемой современной медицинской науки и занимают одно из ведущих мест в структуре младенческой смертности, заболеваемости и ранней детской инвалидности, составляя около 25% от всех врожденных аномалий у детей [2, 3, 10]. Также во многих случаях при наличии выраженных морфологических изменений возникают резистентные эпилептические приступы и грубый неврологический дефицит. Поэтому вопросы своевременной диагностики (в частности, пренатальной), профилактики, лечения и прогнозирования врожденной патологии ЦНС во многих странах мира занимают ведущее направление. При этом необходимо отметить, что врожденные аномалии ЦНС имеют полиэтиологичную, многофакторную природу. Кроме генетических нарушений, определенную роль в их возникновении могут составлять нарушения эмбриогенеза на различных этапах беременности с участием различных экзо- и эндотоксинов, инфекционных факторов (вирус герпеса, цитомегаловирус, токсоплазма и т.д.) [1, 25].
Следует выделить несколько последовательных этапов онтогенеза головного мозга, с каждым из которых может быть связан определенный порок развития головного мозга:
– дорзальная индукция (3–4-я недели гестации; образование нервной трубки, клеток оболочек мозга, каудальных отделов нервной трубки), при нарушении процесса дорзальной индукции основными пороками являются анэнцефалия, энцефалоцеле, аномалия Киари;
– вентральная индукция (5–10-я недели гестации; формирование передних отделов мозга и структуры лица), при нарушении вентральной индукции среди основных пороков ЦНС выделяют голопрозэнцефалию, септооптическую дисплазию, лобарную аплазию, агенезию прозрачной перегородки;
– нейрональная и глиальная пролиферация (2–5-й месяцы гестации; пролиферация нейронов и глии в перивентрикулярных участках), при нарушении нейрональной пролиферации основными пороками являются микролиссэнцефалия, гемимегалэнцефалия, мегалэнцефалия и др.;
– нейрональная миграция (3–5-й месяцы гестации; смещение клеток к периферии и формирование коры и субкортикальных структур, а также формирование слоев коры мозжечка), при нарушении процесса нейрональной миграции основными пороками являются лиссэнцефалия, гетеротопия, агенезия мозолистого тела и др.;
– организация и миелинизация (с 6-го месяця до рождения и постнатального периода; формирование слоев коры, развитие аксонов, дендритов, синапсов), при нарушении организации и миелинизации основными пороками являются полимикрогирия, шизэнцефалия, микродисгенезия.
В первой половине беременности преобладают процессы формирования мозговых структур и миграции нейронов, а во второй половине – начало процессов миелинизации нервных волокон, при этом каждый порок развития имеет связь с определенным периодом развития нервной системы [11, 12, 37]. Данные аспекты необходимо учитывать в процессе пренатальной диагностики врожденных мальформаций ЦНС.
Гемимегалэнцефалия (унилатеральная мегалэнцефалия) – редкий врожденный порок развития ЦНС, характеризующийся диспластическим увеличением одного из полушарий головного мозга в результате аномальной пролиферации нейрональных и глиальных клеток, не имеет различий по расовым и половым признакам и составляет 0,1–0,3% случаев детской эпилепсии. При этом диспластические изменения могут отмечаться и в противоположной гемисфере. Наиболее часто изменения в коре увеличенного пораженного полушария мозга могут быть представлены в виде участков лиссэнцефалии, полимикрогирии, пахигирии, агирии и гетеротопии серого вещества [4, 7, 31]. Данная патология может встречаться в изолированном виде и в сочетании с другими мальформациями ЦНС. Также выделяют три формы гемимегалэнцефалии: изолированную, синдромальную (в составе различных синдромов) и тотальную (включая ипсилатеральную гипертрофию полушария мозжечка, подкорковых ядер, ствола и спинного мозга). Среди ассоциированных с гемимегалэнцефалией синдромов и нейро-кожных аномалий выделяют синдром Клиппеля–Треноне–Вебера (сочетание с гемигипертрофией всего тела), Мак-Кьюна–Олбрайта, эпидермального невуса, протеуса, унилатерального гипомеланоза Ито, нейрофиброматоз 1 типа (Реклингхаузена), туберозный склероз. Четких статистических данных о частоте встречаемости изолированной, синдромальной и тотальной форм гемимегалэнцефалии в настоящее время не существует. По данным Tinkle B.T. и соавт., среди представленных ими ретроспективно 15 случаев гемимегалэнцефалии с 1990 по 2003 г. в 53% (8/15) отмечались несиндромальные формы гемимегалэнцефалии и в 47% (7/15) – синдромальные [13, 17, 29, 33, 35].
Среди основных клинических проявлений гемимегалэнцефалии следует отметить резистентные эпилептические приступы, контралатеральный гемипарез, гемианопсию и выраженные когнитивные нарушения. Могут встречаться макроцефалия, макродонтия, гемифациальная или гемикорпоральная гипертрофия и, в редких случаях, лицевой липоматоз [3, 9, 30]. Эпилептические приступы могут возникать вскоре после рождения, встречаются практически во всех случаях гемимегалэнцефалии и часто носят резистентный характер. Характерна высокая частота приступов, часто статусное течение. Эпиприступы представлены различными типами: фокальные, фокальные с вторичной генерализацией, инфантильные спазмы, миоклонии, тонические, клонические, epilepsia partialis continua, дроп-атаки, атипичные абсансы, бессудорожный эпилептический статус, негативный эпилептический миоклонус. Может отмечаться определенная эволюция патологического процесса: неонатальные судороги (в виде генерализованных клонических пароксизмов и «стертых» приступов – окулоклонус, моргательные движения, подергивания уголка рта, жевательные автоматизмы), затем синдром Отахара (чаще в виде асимметричных тонических спазмов) с последующим переходом в синдром Веста (флексорные и экстензорные асимметричные инфантильные спазмы), затем фокальные приступы в 1–2 года и развитие epilepsia partialis continua в 3–4 года жизни. Для детей школьного возраста характерно преобладание фокальных приступов с усилением выраженности гемипареза. Следует отметить и наличие при гемимегалэнцефалии псевдогенерализованных приступов. Данные пароксизмы могут проявляться в виде фокального эпилептического миоклонуса, атипичных абсансов, негативного миоклонуса, миоклонически-астатических приступов, коротких тонических спазмов [5, 15, 21, 22].
Среди электроэнцефалографических изменений характерна асимметрия основного ритма с преобладанием высокоамплитудной активности на пораженной стороне. Отмечается продолженное региональное или латерализованное замедление и постоянная продолженная региональная или диффузная эпилептиформная активность. У детей в первые месяцы жизни отмечается паттерн «вспышка-угнетение», как правило, асимметричного характера. К 6 месяцам формируется паттерн гипсаритмии, который обычно носит асимметричный характер и регистрируется до года. Отмечается высокоамплитудная активность «острая –медленная волна», возникающая регионально, латерализованно по дефектному полушарию или диффузно, феномен вторичной билатеральной синхронизации. У детей в старшем возрасте диффузные ЭЭГ паттерны могут отсутствовать, а региональная эпилептиформная активность представлена постоянными низкоамплитудными спайками, что характерно для epilepsia partialis continua [5, 27, 36].
Диагноз данной аномалии устанавливается с помощью клинических, электроэнцефалографических данных и методов нейровизуализации (ультразвуковое исследование, компьютерная томография (КТ), магнитно-резонансная томография (МРТ), наиболее информативным из которых является метод МРТ головного мозга. На МРТ изображениях определяются характерные изменения пораженного полушария в виде его увеличения, кольпоцефалия (смещение затылочной доли через срединную линию), нечеткая граница между серым и белым веществом, смещение межполушарной щели в здоровую сторону. Боковой желудочек увеличенного полушария увеличен, а его передний рог выпрямлен и удлинен. В белом веществе мозга могут отмечаться изоинтенсивные серому веществу участки гетеротопии, достигающие больших размеров, в Т2-режиме – гиперинтенсивный сигнал как следствие сочетания глиоза и участков гипомиелинизации. С помощью методов однофотонной эмиссионной томографии и позитронно-эмиссионной томографии в пораженной гемисфере определяются зоны гипометаболизма [6, 7, 16, 31]. При проведении магнитно-резонансной спектроскопии в пораженном полушарии в белом веществе отмечается снижение глутамата, N-ацетиласпартата и креатина с менее выраженными изменениями или отсутствием изменений в сером веществе [23].
Необходимо отметить и возможность пренатальной диагностики гемимегал-энцефалии с помощью пренатальной эхографии и МРТ. Метод пренатальной эхографии (УЗИ) позволяет выявить увеличение одного полушария и ипсилатеральную вентрикуломегалию, однако не позволяет оценить диспластические изменения мозга плода [8, 28]. Также следует отметить, что при проведении метода УЗИ не всегда возможна четкая визуализация исследуемых анатомических структур мозга – из-за рубцовых изменений брюшной стенки, неудобного положения плода, костной тени, избыточного количества амниотической жидкости, многоплодия, ожирения матери и т.д. [18, 19, 38]. В таких ситуациях и при подозрении на врожденную аномалию ЦНС метод МРТ после 20-й недели беременности позволяет значительно улучшить качество пренатальной диагностики, уточнить диагноз и решить вопрос о дальнейшей тактике ведения беременности при отсутствии вредного воздействия на плод. Пренатальной диагностике отводится определяющая роль. Описано и рекомендуется применение метода МРТ для уточнения диагноза, начиная с 18-й недели беременности [20].
Лечение гемимегалэнцефалии представляет собой тяжелую задачу, основная цель которой – контроль над эпилептическими приступами. Устранение врожденного гемипареза и умственной отсталости невозможно. Эпиприступы в неонатальном периоде, склонность к статусному течению, полиморфный характер приступов, формирование контралатеральных очагов эпиактивности в «здоровой гемисфере» по отношению к пораженному полушарию, нарастание когнитивных нарушений и гемипареза являются неблагоприятными прогностическими признаками. Среди существующих методов лечения гемимегалэнцефалии выделяют противосудорожную терапию, кетогенную диету, стимуляцию блуждающего нерва и хирургические методы лечения, среди которых наиболее радикальным и более безопасным методом является функциональная гемисферотомия – с функциональным разобщением пораженного полушария. В случае раннего появления эпиприступов, начиная с неонатального периода, их частого, некупируемого характера, диффузными очагами эпиактивности и выраженным гемипарезом показана ранняя функциональная гемисферотомия, на первом году жизни [14, 26, 32]. Эффект от хирургического лечения при гемимегалэнцефалии более низкий, чем при других формах фокальных кортикальных дисплазий. При гемимегалэнцефалии авторы сообщают о приблизительно 40% эффективности оперативного вмешательства: 38% пациентов в последующем не получали противосудорожное лечение, а при других формах фокальных кортикальных дисплазий эффект от операции составлял 50–70% [14, 24, 34].
За последние 5 лет в клинике детской психоневрологии Института педиатрии, акушерства и гинекологии НАМН Украины проходили обследование и лечение 4 ребенка с гемимегалэнцефалией. Из этих 4 случаев у одного пациента гемимегалэнцефалия сочеталась с гипоплазией мозолистого тела, а у второго – с лиссэнцефалией, левосторонней церебеллярной дисплазией и гипоплазией мозолистого тела соответственно. Эпилептические приступы и выраженная задержка психо-моторного развития были во всех случаях.
Клинический случай. Пациент М., 2 года, поступил в отделение по поводу врожденного порока развития головного мозга – гемимегалэнцефалии в сочетании с лиссэнцефалией, мозжечковой левосторонней дисплазией, гипоплазией мозолистого тела; симптоматической эпилепсией, спастическим тетрапарезом, выраженной задержкой психомоторного развития. Эпилептические приступы были 5–6 раз в неделю, в основном в случае засыпания или просыпания. Характер приступов: асимметричные тонические с напряжением нижних конечностей, вытянутой правой рукой и согнутой левой с последующими клоническими подергиваниями правых конечностей (продолжительность до минуты, могли быть 12–15 раз в день). Также отмечались отведения глазных яблок влево.
Из анамнеза: ребенок от первой беременности, первых родов. Родители ребенка соматически здоровы. Во время беременности: токсикоз первой половины. Обследование на инфекции ТОРЧ-комплекса: результаты отрицательные. При проведении УЗИ в сроке 22–23 и 26–27 недель отмечалось многоводие, «асимметрия в развитии полушарий мозга плода за счет увеличения в размерах левого полушария, расширение бокового желудочка слева до 16,8 мм, левое полушарие мозжечка больше по размерам правого полушария, форма головки плода неправильной формы». В 34 недели беременности проведено МРТ малого таза матери с заключением: МР-признаки гемимегалэнцефалии плода слева. Родоразрешение проведено в сроке 40 недель путем операции кесарева сечения. Масса при рождении 4100 г, длина тела 56 см, окружность головы 33 см. Закричал на 3–4-й минуте, проводились реанимационные мероприятия с последующим пребыванием в реанимационном отделении в течение недели, затем в отделении патологии новорожденных в больнице по месту жительства. Эпилептические приступы и задержка в развитии с рождения. Среди эпилептических приступов отмечались правосторонние гемиконвульсивные приступы, с 4 месяцев – асимметричные тонические спазмы, после года – генерализованные тонико-клонические припадки и ритмичные отведения глазных яблок влево. Периодически – статусное течение, увеличение частоты приступов на фоне инфекции и гипертермии. Перед поступлением в отделение характер приступов в виде асимметричных тонических с последующими клоническими подергиваниями правых конечностей и в виде отведения глазных яблок влево. Из противосудорожных препаратов на момент поступления принимал фенобарбитал (0,01 г три раза в день) и депакин (35 мг/кг массы тела в сутки).
В неврологическом статусе: окружность головы 57 см, асимметрия лица, признаки синдрома Пьера Робена (гипоплазия нижней челюсти, глоссоптоз, высокое готическое нёбо, расщелина верхнего нёба). За предметом не следит. Глазные щели, S<D. Парез n. oculomotorius левого глаза. Тонус в конечностях повышен, больше слева. Сухожильные рефлексы, d<s, спастические. Брюшные рефлексы живые. Двусторонний симптом Бабинского. Опоры нет. Не сидит, не переворачивается. Предметы в руки не берет. Инструкции не выполняет. Слов и звукоподражания нет. Выражает эмоции криком, плачем, усмешкой. Отмечалась выраженная задержка психоэмоционального и статокинетического развития.
Консультация офтальмолога: глазные щели, S<D, оптические среды прозрачны, диски зрительных нервов бледные, границы четкие, резко сужены сосуды. Диагноз: субатрофия зрительных нервов обоих глаз, парез n. oculomotorius левого глаза.
Согласно цитогенетического исследования (материал для исследования – периферическая кровь), у ребенка нормальный мужской кариотип (46, ХУ).
Магнитно-резонансная томография (описание): на серии МР-томограмм головного мозга неравномерное увеличение размеров полушарий большого мозга и мозжечка слева (рис. 1). В лобной и теменной долях левого полушария большого мозга отмечается сглаженность рельефа борозд, кора неравномерно утолщена. Мозолистое тело в передних отделах утолщено, в задних истончено. В белом веществе полушарий большого мозга определяется повышение интенсивности МР сигнала на Т2 ВИ, что обусловлено несовершенной миелинизацией. Базальные ядра слева не дифференцированы. Калибр мозговых артерий слева увеличен по сравнению с правыми. Неравномерно утолщена кора левого полушария мозжечка. Неравномерно гиперинтенсивный МР сигнал от белого вещества левого полушария мозжечка на Т2 ВИ. Дифференциация белое – серое вещество в левых полушариях большого мозга и мозжечка нарушена. Правый боковой, III и IV желудочки деформированы. Затылочная доля полушария большого мозга и задние отделы полушария мозжечка слева смещены в правую сторону за среднюю линию до 1,8 см. Зрительные нервы, хиазма, гипофиз, стволовые отделы мозга без особенностей. Расширены подпаутинные пространства над полушариями большого мозга. Определяется асимметрия размеров лицевого черепа за счет увеличения размеров верхней челюсти слева и значительного увеличения объема подкожной жировой клетчатки. Отмечается уменьшение размеров нижней челюсти, смещение языка к задней стенке глотки и вверх (глоссоптоз), расщелина верхнего неба
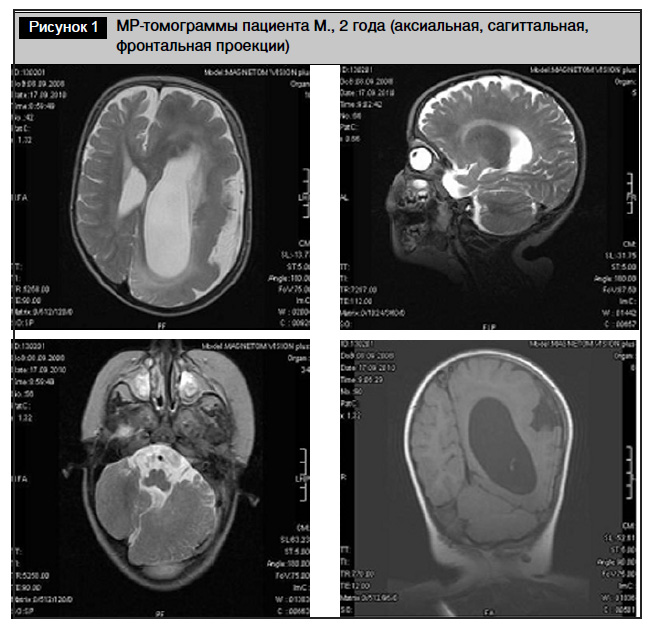
МРТ головного мозга. Заключение: МР-признаки сочетанной конгенитальной мальформации: гемимегалэнцефалии слева, лиссэнцефалии, мозжечковой дисплазии слева, гипоплазии мозолистого тела, аномалии развития лицевого черепа и мягких тканей лица слева, синдрома Пьера Робена.
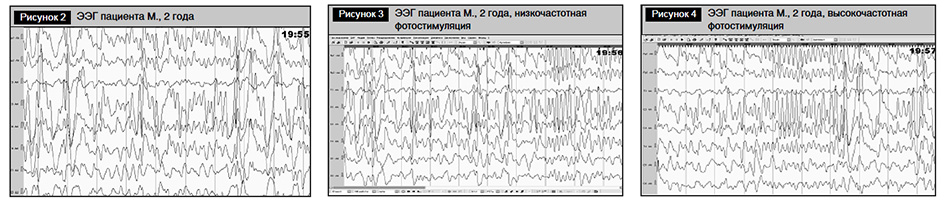
ЭЭГ. Заключение. Паттерн высоковольтной дезорганизованной ЭЭГ с большим количеством полифазных, заостренных волн частотой 4–6 Гц с периодическим замедлением до частоты 2–3 Гц, преимущественно в центрально-височных отведениях, с единичными комплексами острая – медленная волна, пик – медленная волна в левой, центральной и затылочной областях. На фотостимуляцию отсутствовала реакция усвоения ритма, отмечался стойкий очаг патологической активности в левых центральных областях, нарастание высоковольтной дельта-активности в правой височной области (рис. 2–4).
В процессе лечения (нейропротекторная, противосудорожная, метаболическая терапия) была проведена коррекция противосудорожной терапии с отменой фенобарбитала, уменьшением дозы депакина до 30 мг/кг в сутки и назначением топирамата по схеме путем постепенной титрации до 5 мг/кг массы тела в сутки. Отмечено снижение частоты эпилептических приступов до 2–3 в месяц, ребенок стал более спокойным, улучшился сон.
Таким образом, гемимегалэнцефалия является редкой и тяжелой патологией ЦНС с неблагоприятным прогнозом. Она может встречаться как в изолированном виде, так и в сочетании с различными другими аномалиями. В приведенном клиническом случае гемимегалэнцефалия сочеталась с множественными структурными аномалиями в виде лиссэнцефалии, левосторонней мозжечковой дисплазии, гипоплазии мозолистого тела, аномалий развития лицевого черепа и мягких тканей лица слева с признаками синдрома Пьера Робена. В доступных нам литературных источниках мы не встречали описания гемимегалэнцефалии в сочетании с такими множественными структурными аномалиями. Характерными особенностями данного случая были раннее начало эпилептических припадков, их частый полиморфный характер, гемипарез и выраженная задержка психоэмоционального и речевого развития. Проведенная терапия в данном случае дала позитивный эффект в виде снижения частоты эпилептических приступов.
Учитывая вышеизложенное, необходимо акцентировать внимание на свое-временной пренатальной диагностике врожденных пороков ЦНС. В случае нечеткой визуализации, при подозрении на наличие у плода врожденной патологии на основании метода пренатальной эхографии с целью уточнения диагноза и дальнейшей тактики ведения беременности и родов необходимо выполнение МРТ малого таза. При комплексном использовании методов УЗИ и МРТ существенно улучшается качество пренатальной диагностики. Если установлена врожденная патология, в частности патологии ЦНС, проводится пренатальный консилиум для решения вопроса о тактике ведения беременности – сохранении или прерывании до 22-й недели беременности. При поздней диагностике врожденной патологии, отказе от прерывания беременности проводится полное клинико-лабораторное обследование и госпитализация беременной женщины в стационар ІІІ уровня для последующего родоразрешения. Своевременная пренатальная диагностика врожденных аномалий ЦНС и гемимегалэнцефалии в частности, является одной из главных задач современной перинатальной медицины со снижением детской смертности и первичной инвалидности.
Л И Т Е Р А Т У Р А
1. Антонов О.В., Богачева Е.В., Комарова А.А., Антонова И.В. [и др.] // Бюл. сибир. медицины. – 2012. – №3. – С.135–138.
2. Бокерия Л.А., Ступаков И.Н., Зайченко Н.М. // Детская больница. 2003. – №1. – С.7–14.
3. Барашнев Ю.И., Бахарев В.А. Эмбриофетопатии. Диагностика и профилактика аномалий ЦНС и скелета. – М.: Триада-Х. – 2010. – 480 с.
4. Мухин К.Ю., Малинина Е.В., Чадаев В.А. // Вестн. СПб. гос. мед. академии. – 2006. –№1. –С.133–137.
5. Мухин К.Ю., Петрухин А.П., Холин А.А. Эпилептические энцефалопатии и схожие синдромы у детей. – М., 2011. – 680 с.
6. Трофимова Т.Н., Ананьева Н.И., Назинкина Ю.В., Карпенко А.К., Халиков А.Д. Нейрорадиология. – СПб.: СПбМАПО, 2005. – 288 с.
7. Abdel Razek A.A., Kandell A.Y., Elsorogy L.G. // Am. J. Neuroradiol. – 2009. – Vol.30 (1). – Р.4–11.
8. Alvarez R.M., Garcia-Diaz L., Mаrquez J. et al. // Fetal. Diagn. Ther. – 2011. – Vol.30 (3). – Р.234–238.
9. Aydingoz U., Emir S., Karli-Oguz K. Kose G. et al. // Pediatr. Radiol. – 2002. – Vol.32 (2). – Р.106–109.
10. Barkovich A.J. Congenital Malformations of the brain and skull. In: Pediatric Neuroimaging (4th edn). Philadelphia: Lippincott Williams & Wilkins; 2005. – Р. 291–439.
11. Barkovich A.J., Kuzniecky R.I., Jackson G.D. et al. // Neurology. – 2001. – Vol.57. – P.2168–2178.
12. Barkovich A.J., Kuzniecky R.I., Jackson G.D. et al. // Neurology. – 2005. – Vol.65. – P.1873–1887.
13. Di Rocco С., Battaglia D., Pietrini D. et al. // Child’s Nervous System. – August 2006. – Vol.22, iss.8. – Р.852–866.
14. Di Rocco C., Iannelli A. Hemimegalencephaly and intractable epilepsy: complications of hemispherectomy and their correlations with the surgical technique. A report on 15 cases // Pediatr. Neurosurg. – 2000. – Vol.33 (4). – P.198–207.
15. Dobyns W.B., Kuzniecky R. Malformation of cortical development and epilepsy. In: The treatment of epilepsy / ed. Wyllie. – 2006. – Р.37–53.
16. Flores-Sarnat L. // J. Child. Neurol. – 2002. – Vol.17. – Р.373–384.
17. Galluzzi P., Cerase A., Strambi M. et al. // J. Child. Neurol. – 2002. – Vol.17. – Р.677–680.
18. Glenn O.A., Barkovich J. // AJNR. – 2006. – Vol.27. – Р. 1604–1611.
19. Glenn O.A., Barkovich J. // AJNR. – 2006. – Vol.27. – Р. 1807–1814.
20. Griffiths P.D., Porteous M., Mason G. et al. // Brit. J. Radiology. – 2012. – Vol.85. – P.e1038–e1045.
21. Guerra M.P., Cavalleri F., Migone N. et al. // J. Child. Neurol. – 2007. – Vol.22 (1). – Р.80–84.
22. Guzzetta F. // Epilepsia. – 2002. – Vol.43 (9). – Р.1106–1109.
23. Hanefeld F. // Epilepsia. – 1995. – Vol.36 (12). – Р.1215–1224.
24. Jonas R. // Neurology. – 2004. – Vol.62 (10). – P.1712–1721.
25. Kinsman S.L., Johnston M.V. Congenital Anomalies of the Central Nervous System. In: Nelson textbook of paediatrics (Kliegman M. et al. eds.18 th ed. Philadelphia: Saunders), 2007. – Р.2443–2448.
26. Maehara T., Shimizu H., Shigetomo R., Tamagawa K. // No To hattatsu. – 2000. – Vol.32. – P.395–400.
27. Ohtsuka Y, Ohno S, Oka E. // Pediatric Neurology. – 1999. – Vol.20 (5). – Р.390–393.
28. Romero X.C., Molina F.S., Pastor E., Amaya F. // Fetal Diagn. Ther. – 2011. – Vol.29 (3). – Р.257–260.
29. Salamon N., Andres M., Chute D.J. et al. // Brain. – 2006. – Vol.129. – Р.352–365.
30. Sasaki M. // J. Child. Neurol. – 2005. – Vol.20 (4). – Р.337–341.
31. Simon D. Shorvon S.D., Andermann F., Guerrini R. The Causes of Epilepsy: Common and Uncommon Causes in Adults and Children Cambridge Univ. Press, 2011. – 787 р.
32. Smith J.R., Fountas K.N., Lee M.R. // Childs Nervous System. – 2005. – Vol.21 (6). – P.466–472.
33. Tagawa T., Futagi Y., Arai H. et al. // Pediatr. Neurol. – 1997. – Vol.17 (2).– Р.180–184.
34. Terra-Bustamante V.C., Machado H.R., Sakamoto A.C. // J. Epilepsy Clin. Neurophysiol. – 2006. – Vol.12 (2). – P.99–105.
35. Tinkle B.T., Schorry E.K., Franz D.N. // Am. J. Med. Genet. 2005. – Vol.139 (Part A.3). – Р.204–211.
36. Vigevano F., Fusco L., Granta T. // In: Dysplasias of cerebral cortex and epilepsy / Eds.: R.Guerrini, F.Andermann, R.Canapicchi. – Philadelpfia: Lippincott-Raven Publishers, 1996. – P.285–294.
37. Volpe J.J. Neurology of the Newborn. Elsevier Health Sciences, 2008. – 1094 р.
38.Wang G.B., Shan P.Q., Ma Y.X. et al. // Chin. Med. J (Engl). – 2006. – Vol.119. – Р.1272–1277.
Медицинские новости. – 2014. – №6. – С. 61-65.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.