В 2003 г. в Рекомендациях Европейского общества по артериальной гипертензии – Европейского общества по кардиологии (ЕОАГ-ЕОК) впервые появились две принципиально новые позиции, касающиеся положения стратификации риска [12]. В частности, в них указывалось, что все больные артериальной гипертензией (АГ) должны быть классифицированы с учетом общего кардиоваскулярного риска, а его стратификация должна строиться на основе выявления субклинических, т. е. ранних признаков поврежденных органов-мишеней.
В этой связи основная цель лечения АГ была пересмотрена. Теперь она заключается в обеспечении максимально возможного снижения риска сердечно-сосудистых осложнений и предполагает не только снижение артериального давления (АД) до целевого уровня, но и коррекцию всех модифицируемых факторов риска и лечение сопутствующих заболеваний. Исходя из этого, максимальный эффект при лечении АГ может быть достигнут только при одновременном влиянии на все три составляющие индивидуального риска развития сердечно-сосудистых заболеваний (ССЗ).
В рекомендациях ЕОАГ-ЕОК 2003 г. также указывалось, что уровень АД в плечевой артерии может быть выше такового в нисходящей аорте (центральное АД), и различные классы антигипертензивных препаратов по-разному влияют на центральное АД в аорте при сопоставимом эффекте влияния на уровень АД в плечевой артерии. Кроме того, были обозначены такие параметры центральной гемодинамики, как скорость аугментации и скорость пульсовой волны, которые могли быть использованы в качестве критериев субклинической оценки повреждения органов-мишеней. Вместе с тем в Рекомендациях ЕОАГ-ЕОК 2003 г. указывалось, что для этого требуется проведение проспективных клинических исследований по изучению прогностической значимости центрального АД и параметров центральной гемодинамики.
В течение нескольких последующих лет результаты целого ряда исследований показали, что центральное АД является наиболее чувствительным индикатором нагрузки на левый желудочек [1, 39]. Кроме того, установлено, что центральное АД по сравнению с периферическим является наиболее значимым фактором в плане оценки риска развития неблагоприятных сердечно-сосудистых событий, так как зависит от эластичности артериальной сосудистой стенки [41]. С другой стороны, появление в клинике неинвазивных методов оценки центральной гемодинамики (контурный анализ пульсовой волны, плетизмография, сфигмография и т.д.) дало возможность получать полную информацию о структурно-функциональном состоянии сосудов [20].
Одновременно ряд крупных исследований последнего десятилетия доказал, что увеличение жесткости артерий – это независимый предиктор развития ССЗ и сердечно-сосудистой смертности [19, 25, 49]. Установлено, что ее прогностическая значимость высока на доклинических стадиях развития заболеваниях [40]. С учетом накопленной информации в Рекомендациях ЕОАГ-ЕОК 2007 г. [13] и их пересмотра в 2009 г. [22] параметры жесткости сосудов были включены в число тестируемых при поиске субклинического поражения органа-мишени при АГ, а также факторов, влияющих на прогноз АГ. По сути, данные изменения Рекомендаций ЕОАГ-ЕОК отражают общемировой уровень признания важной роли сосудов, являющихся одним из главных органов-мишеней при АГ.
Структурно-функциональные изменения сосудистой стенки играют ключевую роль при установлении прогноза различных заболеваний, в том числе АГ. Прогностическая значимость упруго-эластических свойств артерий при артериальной гипертензии тесно связана с процессом ремоделирования сосудов, который способствует нарушению их основных функций: проводящей и демпфирующей [8, 32, 33].
Проводящая функция заключается в обеспечении доставки адекватного количества крови к периферическим тканям в соответствии с их потребностями, что определяется шириной просвета сосуда. Нарушение проводящей функции связано с сужением или окклюзией сосуда и приводит к нарушению перфузии тканей ниже места окклюзии. Проводящая функция позволяет поддерживать на постоянном уровне величину среднего АД между аортой и периферическими артериями, которая в свою очередь зависит от сердечного выброса (СВ) и общего периферического сопротивления сосудов (ОПСС).
Демпфирующая функция обеспечивается эластическими свойствами артерий и направлена на гашение колебаний давления крови, создаваемого сердцем. Так что демпфирование обеспечивает передачу относительно стабильного давления крови периферическим тканям. Данный процесс происходит следующим образом. Как известно, кровоток в аорте и крупных артериях пульсирует в результате ритмичного опорожнения левого желудочка (ЛЖ). При изгнании крови из ЛЖ в систолу эта порция толкает объем крови, уже находящийся в восходящей аорте, создавая при этом волну давления, которая быстро (со скоростью 4–10 м/с) проводится до уровня артериол (рис. 1). Когда пульсовая волна (ПВ) проходит по аорте и крупным сосудам, она создает преходящий градиент давления, способствующий продвижению крови вперед и формирующий пульсирующую кривую потока крови. Именно поэтому кровь проводится вперед короткими толчками, разделенными периодами стаза, а средняя скорость движения крови равна 0,2 м/с [33].
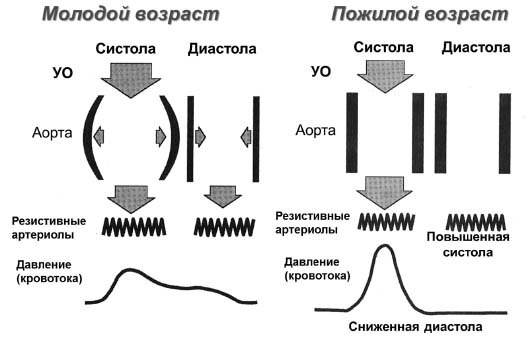
Рисунок 1. Схематическое изображение патогенеза возрастных изменений формы пульсовой волны. Адаптировано из Izzo J.L. [16]
Кривая давления ведет к выпячиванию эластичной артериальной стенки, накапливая, таким образом, энергию проходящей волны. При этом в случае систолического расширения аорты в ней может накапливаться до 40–50% объема СВ и около 10% энергии сердечного сокращения. Затем артериальная стенка возвращается к прежнему состоянию, высвобождая часть энергии и продвигая кровь вперед во время диастолы (диастолический поток).Этот насосный механизм эластичных артерий обозначают термином «функция Windkessel». Большие артерии также кумулируют и используют часть энергии кривой давления. Пульсирующий характер потока прогрессирующе снижается. Однако пульсовое артериальное давление (ПАД) становится больше по мере уменьшения диаметра артерий, вплоть до мелких, где происходит постепенное исчезновение этой волны. Поэтому по мере удаления от сердца, т. е. продвижения от восходящей аорты к периферии наблюдается увеличение ПАД (рис. 2). Отчасти это связано с тем, что часть фракции кривой давления возвращается к сердцу от всех зон артериальной бифуркации. При этом формируется волна отражения[28].
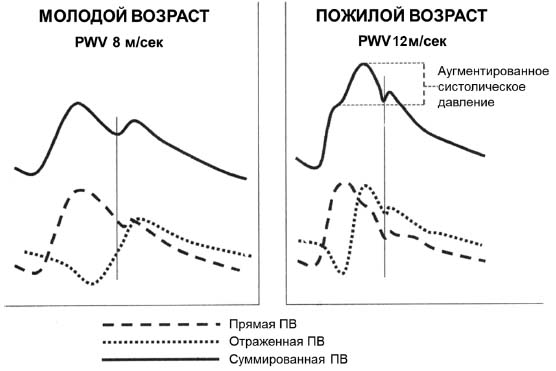
Рисунок 2. Влияние возраста на изменение контура пульсовой волны. Адаптировано из Franclin S.S., Izzo J.I. [9]
Как известно, систолическое артериальное давление (САД) определяется величинами СВ, ОПСС и упруго-эластическими свойствами крупных сосудов (аортально-артериальной жесткостью) и возрастает, если увеличивается любой из этих показателей [26]. Величина диастолического артериального давления (ДАД) определяется ОПСС и упруго-эластическими характеристиками аорты, от которых зависит величина «возвращенной» в кровоток демпфированной во время систолического расширения крови. Установлено, что ОПСС и упруго-эластические свойства артерий, которые характеризуются жесткостью сосудистой стенки, оказывают прямо противоположное влияние на величину ДАД. Так, с увеличением ОПСС наблюдается рост ДАД, а с увеличением жесткости артерий – его снижение. АД в условия диастолы прогрессирующе снижается, так что укорочение диастолического интервала, связанного с увеличением частоты сердечных сокращений (ЧСС) также повышает ДАД. В этих условиях ПАД, представляющее собой разницу САД и ДАД, будет увеличиваться. Таким образом, жесткость артерии является важным параметром, определяющим величины САД, ДАД и ПАД [20].
Жесткость артерии напрямую связана со скоростью распространения ПВ – Pulse Wave Velocity (PWV), являющейся «золотым стандартом» измерения жесткости артерий и независимым предиктором развития ССЗ, а также общей сердечно-сосудистой смертности [14]. При этом увеличение скорости ПВ на 1 м/с увеличивает риск смерти на 10%.
Жесткость сосудистой стенки зависит от выраженности атеросклеротических изменений, скорости и степени возрастной инволюции структурных белков эластина и фибулина, возрастного повышения жесткости коллагена, а также локализации сосудов [15, 26]. В сосудистой стенке периферических артерий содержание коллагена выше по сравнению с центральными сосудами, что делает их более жесткими [15].
Возраст – это важнейший фактор риска развития неблагоприятных сердечно-сосудистых событий, что в значительной степени связано с увеличением жесткости артерий [26, 27]. У лиц молодого возраста отраженная волна поступает в раннюю диастолу, что способствует приросту ДАД и улучшению, таким образом, коронарного кровотока. С возрастом наблюдается повышение жесткости артерии и увеличение скорости ПВ. Это приводит к тому, что отраженная волна, возвращаясь в аорту значительно быстрее, чем в норме, попадает в позднюю систолу, в результате чего наблюдается увеличение САД. Из-за повышения жесткости сосудистой стенки одновременно с повышением САД и ускорением отраженной волны наблюдается снижение ДАД. В то же время ПАД увеличивается. В результате повышается постнагрузка на ЛЖ, развивается гипертрофия миокарда, ухудшается коронарная перфузия миокарда и нарушается диастолическая функция ЛЖ.
Повышение жесткости крупных сосудов при старении в значительной степени обусловлено развитием артериосклероза, который необходимо отличать от атеросклероза [31]. Артериосклероз следует считать дегенеративным процессом, вследствие развития которого эластические волокна, находящиеся в медиальном слое сосудистой стенки, постепенно замещаются коллагеновыми. Данные структурные изменения приводят к ремоделированию сосудов и нарушению их эластических свойств. Они становятся дилатированными и приобретают патологическую извитость. По понятным причинам в этих условиях страдают демпферные свойства крупных сосудов.
В отличие от артериосклероза, в основе атеросклероза лежит иммуновоспалительный процесс, локализующийся в интиме. На начальных этапах заболевания образуется фиброатерома, а в развернутой стадии атеросклероз проявляется окклюзией, что нарушает проводящую функцию сосудов. Однако при всей несхожести этих двух заболеваний, по сути, они являются лишь разными формами структурного ответа на метаболические и гемодинамические повреждения. Общим пусковым фактором этих двух процессов является нарушение функции эндотелия, регулирующего высвобождение вазоконстрикторов и вазодилататоров и повышение тонуса сосудов [43]. Данная дисфункция вызывает гипертрофию и гиперплазию гладкомышечных клеток сосудистой стенки, активацию синтеза соединительного матрикса, утолщение медии артерий. В сумме все это приводит к нарушению упруго-эластических свойств сосуда. В то же время, гипертрофия гладкомышечных клеток увеличивает степень вазоконстрикции в ответ на нейрогормоны, приводит к повышению ОПСС и развитию артериальной гипертензии [42].
Таким образом, жесткость сосудов – это интегральный показатель, определяемый не только структурными элементами сосудистой стенки и давлением крови, но также и регуляторными механизмами, среди которых эндотелиальная дисфункция играет ключевую роль.
Популяционные исследования свидетельствуют о разнонаправленных изменениях САД и ДАД в различные годы жизни [10]. Так, САД начиная с юношеского возраста и в течение всей последующей жизни, неуклонно возрастает. По другой траектории изменяется ДАД. До достижения человеком 50-летнего возраста значение ДАД увеличивается, а начиная с 60 лет снижается. Соответственно, в старшем возрастном периоде увеличивается и ПАД.
Возрастные изменения АД находятся в прямой зависимости от возрастной динамики ОПСС и жесткости сосудов. При этом различают три основных возрастных периода: до 45–50 лет, 50–60 лет и старше 60 лет. В молодом возрасте, когда еще нет значимых изменений сосудистой стенки, основные механизмы повышения АД – дисфункция эндотелия и гипертрофия гладкомышечных клеток резистивных сосудов. Соответственно, в этом возрасте величина АД будет зависеть преимущественно от уровня ОПСС. В возрасте 50–60 лет, когда сосуды уже теряют эластичность, величина АД определяется ОПСС и жесткостью сосудов в равной мере. В результате ДАД мало изменяется, в отличие от величины САД, которая продолжает увеличиваться. У лиц в возрасте 60 лет и старше за счет развития артериосклероза и атеросклероза наблюдается процесс значимого увеличения жесткости преимущественно крупных сосудов, который является определяющим в снижении ДАД и, соответственно, в еще большем росте САД, а также ПАД.
Изменение уровня АД связано с риском развития сердечно-сосудистых осложнений. Однако в зависимости от возраста САД и ДАД имеют разную прогностическую значимость [11]. В молодом возрасте, до 50 лет, риск развития неблагоприятных сердечно-сосудистых осложнений напрямую зависит от уровня ДАД, а в более пожилом возрасте – от уровня САД. В последнее время установлено, что еще большей прогностической значимостью обладает величина ПАД, в особенности в аорте даже на субклинической стадии развития атеросклероза [38]. Доказано, что упруго-эластические свойства крупных артерий имеют более значимый вклад в развитии риска сердечно-сосудистых осложнений, чем ОПСС.
Периферическое сосудистое сопротивление прогрессивно возрастает по мере удаления от сердца из-за естественного прогрессивного сужения просвета артерии, поэтому в норме САД выше в периферических артериях, чем аорте [32]. В то же время ДАД, хотя и незначительно, но снижается, что связано с небольшой величиной отраженной от бифуркации аорты волны на верхних и нижних конечностях, по сравнению с отраженной волной в аорте. В плечевой артерии ДАД уменьшается на ~1 мм рт. ст. В результате САД и ПАД выше на ногах и руках, чем в нисходящей аорте. Повышение САД зависит от возраста, частоты сердечных сокращений, положения тела, физической нагрузки, приема вазодилататоров.
Степень повышения САД на периферии по сравнению с аортой зависит от упруго-эластических свойств артерий и удаленности места измерения. В этой связи манжеточное давление в плечевой артерии далеко не всегда соответствует давлению в нисходящей аорте [1]. Поскольку величина САД в аорте, в основном, определяет постнагрузку и величину массы миокарда ЛЖ [38], которые являются независимыми предикторами сердечно-сосудистой смертности, то корреляция между уровнем САД на плече и показателями смертности имеет более опосредованный характер по сравнению с САД в аорте [34, 36].
Установлено, что центральное АД является наиболее чувствительным индикатором повреждения органа-мишени, а также риска различных сердечно-сосудистых заболеваний не только у пациентов с атеросклерозом [47], но и у здоровых лиц [29]. При этом центральное САД отражает нагрузку на ЛЖ и тесно коррелирует с индексом массы ЛЖ, независимо от возраста и уровня среднего АД [38], а САД в сонной артерии – с толщиной стенки ЛЖ [37]. Более того, величина центрального САД зависит от структурно-функциональных свойств сосудистой стенки. Выявлена связь между величиной САД в аорте и степенью гипертрофии сосудистой стенки, выраженностью атеросклероза в сонной артерии [4, 36], диаметром аневризмы восходящей аорты при синдроме Марфана [17].
Таким образом, сосуды и их упруго-эластические свойства играют значимую роль в формировании АГ. Это вывод принципиально важен, так как отсюда следует, что лечение АГ должно быть направлено не только на восстановление функции резистивных, но и крупных эластических артерий, которые следует рассматривать в качестве органа-мишени. При этом эффективность антигипертензивной терапии во многом должна определяться влиянием на процесс уплотнения крупных сосудов.
Подтверждением этого положения является исследование ASCOT [44]. Оно было посвящено сравнительной оценке эффективности профилактики осложнений ССЗ при лечении АГ, основанном на применении атенолола в сочетании с бендрофлуметиазидом (стандартный режим) и норваска в сочетании с периндоприлом (современный режим). Основная цель состояла в изучении влияния двух разных схем антигипертензивной терапии на частоту развития нефатального инфаркта миокарда (ИМ) с клиническими проявлениями и без них и смертность от ишемической болезни сердца (ИБС).
В исследовании участвовали 19 257 пациентов в возрасте от 40–79 лет с нелеченой АГ (САД 160 мм рт. ст. и/или ДАД 100 мм рт. ст.) или леченой АГ (САД 140 мм рт. ст. и/или ДАД 90 мм рт. ст.) при наличии не менее трех дополнительных факторов риска. К этим факторам риска относились: гипертрофия ЛЖ по критериям Американского общества эхокардиографии, Корнельского произведения или индекса Соколова–Лайона, наличие патологических отклонений на ЭКГ (патологический зубец Q, блокада левой ножки пучка Гиса, изменения сегмента ST или зубца Т ишемического генеза), сахарный диабет 2-го типа (не требующий инсулинотерапии), поражение периферических артерий, наличие цереброваскулярных событий в анамнезе (включая транзиторные ишемические атаки) не менее чем 3 мес. тому назад, мужской пол, возраст старше 55 лет, микроальбуминурия или протеинурия, регулярное курение, отношение концентрации общего холестерина и холестерина высокой плотности в плазме крови 6, ранее (до 55 лет у мужчин и до 60 лет у женщин) развитие ИБС у родственников первой линии.
Больных случайным образом распределяли на две группы. В первой группе (n=9639) пациенты получали блокатор кальциевых каналов (БКК) норваск (амлодипин, Pfizer) с добавлением ингибитора ангиотензинпревращающего фермента (иАПФ) периндоприла в случае необходимости усиления гипотензивного эффекта, во второй (n=9618) – β-адреноблокатор (БАБ) атенолол с добавлением тиазидного диуретика (ТД) бендрофлуметиазида и калия. Начальная доза норваска была 5 мг/сут, при необходимости ее увеличивали до 10 мг/сут. Начальная доза атенолола составила 50 мг/сут, при необходимости ее увеличивали до 100 мг/сут. В случае комбинированной терапии дозировка периндоприла была в пределах 4–8 мг/сут, а бендрофлуметиазид назначался в дозе 1,25–2,5 мг/сут. В случае отсутствия достижения желаемого гипотензивного эффекта в обеих группах к терапии назначался β-блокатор доксазозин в дозе 4–8 мг/сут. К концу исследования выяснилось, что в 77,8% случаев больные получали комбинированную терапию. При этом в группе современного лечения в 68,4% случаев использовали комбинацию норваск + периндоприл, а в группе стандартного лечения в 55,7% – комбинацию атенолол + бендрофлуметиазид.
В 2004 г., примерно через 5,4 года от начала исследования, в соответствии с рекомендациями Наблюдательного комитета по безопасности, исследование было прекращено досрочно, поскольку было выявлено статистически значимое различие между двумя группами почти по всем основным и дополнительным показателям [5]. Кроме того, дальнейший прием стандартной схемы лечения увеличивал вероятность развития неблагоприятных исходов у пациентов по сравнению с современной схемой лечения.
В обеих группах наблюдалось снижение САД и ДАД. Среднее значение АД в группе БКК+иАПФ составило 136,1/77,4 мм рт. ст., а в группе БАБ+ТД – 137,7/79,2 мм рт. ст. Таким образом, в группе, где использовалась комбинация норваск+периндоприл, значения АД были на 2,7/1,9 мм рт. ст. ниже, чем в группе, где применяли атенолол+ТД. В этой же группе частота нефатального ИМ и фатальной ИБС (первичная конечная точка) была на 10% ниже, чем в группе атенолола и бендрофлуметиазида, хотя она и не была достоверной. Однако установлено значительное снижение частоты развития различных неблагоприятных событий, которые оценивали по вторичным точкам. Так, на 11% уменьшилась частота развития смертности от всех причин, на 13% – всех случаев смерти от ИБС, на 16% – смертности от ССЗ, на 23% снизилась частота развития смертельного и несмертельного инсульта, а на 14% – частота развития сердечной недостаточности.
Поиск причин для столь существенного уменьшения частоты развития неблагоприятных исходов в группе норваска показал, что такое незначительное различие в уровне АД между двумя группами может объяснить только 4–8% от полученного снижения коронарного риска и 8–14% от полученного снижения риска инсульта. Была попытка связать полученные результаты с неблагоприятным влиянием БАБ и ТД на углеводный и жировой обмен. Однако, по данным мультивариантного анализа изменения биохимических показателей крови, а также таких параметров, как ЧСС и индекс массы тела, могут объяснить различия по коронарным событиям на 50%, а по инсультам – на 40% от полученных значений снижения в группе норваска [35].
Возможность участия принципиально других факторов, способствующих достоверному различию в конечных исходах, наблюдаемых в исследовании ASCOT-BPLA, была обнаружена при анализе результатов следующих исследований: CAFЙ [50], CAFЙ-Heart RATE [53] и ASCOT-LLA [45]. В рамках программы исследования ASCOT-BPLA проведено субисследование CAFЙ, в котором была дана сравнительная оценка влияния норваска и атенолола на центральную и периферическую гемодинамику. К моменту проведения данного исследования значимость уровня АД в плечевой артерии, как фактора риска развития смерти и заболеваемости от ССЗ уже была многократно установлена в различных исследованиях и метаанализах [48, 52, 54]. В то же время влияние уровня АД в аорте на развитие неблагоприятных сердечно-сосудистых событий было неизвестно.
В субисследование CAFE были включены более 2000 больных, ранее участвовавших в исследовании ASCOT-BPLA. Всем пациентам в течение 5 лет наблюдения периодически проводили измерение АД в плечевой артерии тонометрическим методом и инвазивным методом в аорте. Одновременно изучали изменение гемодинамических параметров. Результаты показали, что между группами норваска и атенолола отсутствуют достоверные различия в снижении САД и изменении величины пульсового АД в плечевой артерии. В то же время по сравнению с группой атенолола в группе норваска наблюдалось достоверное снижение АД в аорте. В частности, различие между группами в уровне САД составило 4,3 мм рт. ст. (3,3–5,4; p<0,0001), пульсового АД – 3,0 (2,1–3,9; p<0,0001).
Объяснение причин более выраженного снижения САД и ПАД в аорте в группе больных, получавших норваск+периндоприл, были получены при анализе динамики ПВ. Установлено уменьшение прямой ПВ на фоне приема БАБ+ТД, что было связано со снижением СВ. В то же время наблюдалось увеличение амплитуды отраженной волны, что отражало повышение уровня ОПСС. Известно, что наибольший вклад в формирование САД имеет отраженная волна. Соответственно, на фоне приема БАБ+ТД уровень САД повышался, несмотря на снижение прямой ПВ. В этих же условиях ускорение возврата отраженной волны способствовало снижению ДАД, в результате чего наблюдалось повышение ПАД в аорте. Прямо противоположные изменения контура ПВ наблюдались у больных, принимавших норваск+периндоприл, что говорит о повышении эластичности сосудистой стенки и уменьшении ОПСС на фоне приема норваска и иАПФ.
В исследовании CAFE-Heart RATE [53] установлено, что ЧСС является наиболее мощным фактором, вызывающим различия между уровнем САД и отраженной волной в аорте на фоне приема норваска по сравнению атенололом. Это обусловлено несколькими факторами. Известно, что снижение ЧСС способствует увеличению продолжительности периода изгнания и не оказывает значимого влияния на скорость ПВ [3]. А это значит, что в таких условиях возрастает вероятность попадания отраженной волны в позднюю систолу независимо от скорости ПВ. Таким образом, уменьшение ЧСС на фоне приема БАБ способствует повышению уровня центрального давления, а не периферического и ухудшению, таким образом, гемодинамического профиля (рис. 3).
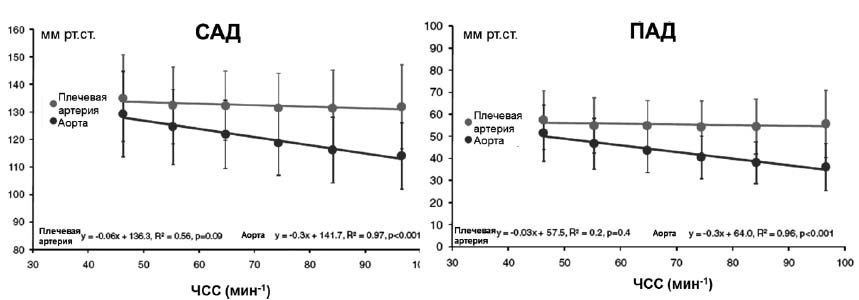
Рисунок 3. Взаимосвязь между ЧСС и САД, ПАД в аорте и плечевой артерии. Адаптировано из Williams B., Lacy P.S. [53]
Кроме того, известно, что уровень давления зависит от ударного объема (УО) и ЧСС [2]. Соответственно, отсюда также прослеживается взаимосвязь между ЧСС и давлением. К примеру, если происходит уменьшение ЧСС, то оно сопровождается компенсаторным увеличением УО. Однако при отсутствии патологических изменений сосудистой стенки, как это бывает у молодых, повышение УО компенсируется вазодилатирующей реакцией. В результате центральная гемодинамика существенно не изменяется. Однако у гипертоников или пожилых людей, в условиях снижения упруго-эластичных свойств артерий и отсутствия должной вазодилатирующей реакции, наблюдается повышение центрального давления в отсутствие компенсаторного снижения ОПСС. По такому же принципу работают и БАБ, прежде всего способствуя снижению ЧСС и вызывая вазоконстрикцию [51].
Анализируя результаты целого ряда исследований и субисследований, можно сделать вывод, что причины наблюдаемых различий в уровне САД и ПАД в аорте на фоне приема норваска по сравнению с атенололом могут быть связаны с отсутствием замедления ЧСС, улучшением упруго-эластических свойств крупных артериальных сосудов, а также достижением ПВ более дистальных отделов артерий вследствие вазодилатации.
Взаимосвязь замедления ЧСС на фоне приема атенолола и повышения центрального давления, установленная в результате субисследования CAFЙ-Heart RATE [53], подняла вопрос для обсуждения антигипертензивного действия других БАБ. По-видимому, в этом случае все же идет речь о единой для всех БАБ физиологической реакции уменьшения ЧСС с соответствующими последствиями. Ведь установлено, что обусловленное БАБ снижение ЧСС повышает центральное давление и отраженную волну даже в условиях дополнительного приема мощных вазодилататоров [46]. Правда, имеется другая точка зрения, основанная на результатах проведенных ранее очень небольших исследований, в которых продемонстрировано, что БАБ с вазодилатирующими свойствами более значимо снижают центральное давление по сравнению БАБ без этих свойств [7, 18]. Однако вряд ли она может рассматриваться всерьез из-за отсутствия большой доказательной базы.
Способность гипотензивных препаратов снижать АД не только за счет снижения скорости ПВ, но и улучшения эластических свойств артерий представляется достаточно важной.
Сравнительной оценке влияния различных классов антигипертензивных препаратов на эластичность сосудистой стенки посвящены несколько исследований [21, 30]. Результаты одного из наиболее значимых были опубликованы в 2004 г. [30]. Целью данного исследования было сравнительное изучение влияние четырех основных классов антигипертензвных препаратов – БКК, ТД, иАПФ, БАБ – на свойства сосудистой стенки (жесткость), которые оценивали по изменению центрального давления в аорте и центрального индекса аугментации (рис. 4). Установлено, что БКК и диуретики снижают САД, как в плечевой артерии, так и в аорте значимо больше, чем БАБ и иАПФ. При этом на фоне применения БАБ увеличение САД в аорте было больше по сравнению с остальными группами препаратов. Так, в группе плацебо увеличение САД и индекса аугментации в аорте составило 23 мм рт. ст. и 33,3% соответственно. На фоне применения различных препаратов данные параметры достигали следующих значений: при применении ИАПФ – 18 мм рт. ст. и 30%, БАБ – 26 мм рт. ст. и 38,5%, БКК – 16 мм рт. ст. и 28%, диуретиков – 17 мм рт. ст. и 28,8%.
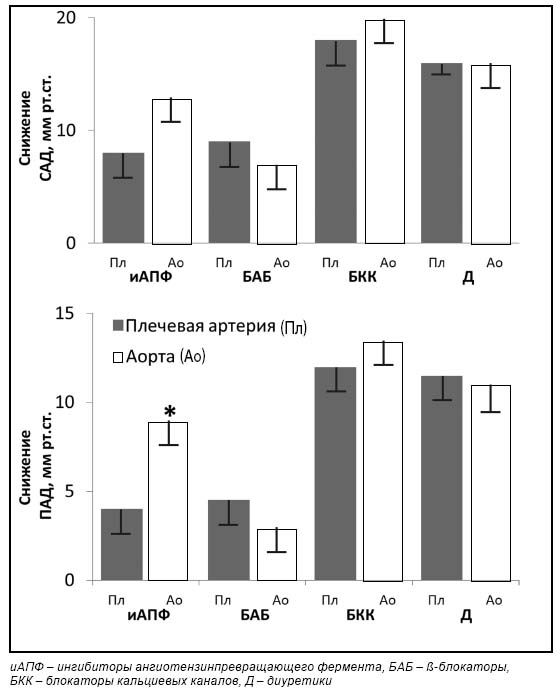
Рисунок 4. Различия между САД и ПАД в плечевой артерии (Пл) и аорте (Ао) на фоне приема основных групп антигипертензивных препаратов. Адаптировано из Wright J.T. et al. [30]
Среди возможных механизмов, обеспечивающих различие в конечных исходах исследования ASCOT-BPLA, рассматривается антиатеросклеротический эффект норваска (амлодипина, Pfizer) [24]. В исследовании PREVENT было установлено благоприятное влияние норваска на прогноз больных ИБС [23]. При систематическом приеме норваска в течение трех лет снизилось число госпитализаций, обусловленных обострением стенокардии и хронической сердечной недостаточности, частота случаев нестабильной стенокардии снизилась на 33%, а число операций по реваскуляризации миокарда – на 43%. Кроме того, прием норваска позволил уменьшить толщину комплекса интима-медиа сонных артерий. Суммарно все эти данные свидетельствует о благотворном влиянии норваска на гладкомышечную мускулатуру или эндотелиальную функцию артериальных сосудов. В последующем экспериментально было установлено, что антиатерогенный эффект норваска многократно усиливается при совместном приеме с аторвастатином, так как оба препарата оказывают лечебное воздействие на разные молекулярные и клеточные механизмы развития атеросклероза [6].
Данное положение было подтверждено результатами субисследования ASCOT-LLA, в котором изучено влияние аторвастатина (липримара, Pfizer) в дозе 10 мг/сут на частоту развития несмертельного ИМ и смертельной ИБС у больных АГ, принимавших участие в исследовании ASCOT-BPLA [45]. Установлено статистически значимое различие по всем основным и дополнительным показателям между группами пациентов, принимавших липримар или плацебо. В частности, в группе больных, принимавших липримар, частота развития несмертельного ИМ и смертельной ИБС снизилась на 36%. При этом в случае сочетанного приема норваска и ИАПФ с липримаром снижение первичной конечной точки по сравнению с плацебо достигало еще более значительной цифры– 53% (p=0,0001). Интересно отметить, что в условиях приема атенолола/бендрофлуметиазида с аторвастатином снижение риска развития неблагоприятных сердечно-сосудистых событий не имело статистической значимости и составило 16% (p=0,025).
Таким образом, возрастные изменения сосудистой стенки крупных артерий, связанные с развитием, прежде всего, артериосклероза, способствуют увеличению их жесткости. В конечном итоге следствием данного патологического процесса является развитие изолированной систолической гипертензии, являющейся преимущественной формой АГ у лиц старше 50 лет. В этой связи способность антигипертензивных препаратов снижать АД в сочетании с улучшением упруго-эластических свойств артерий должна рассматриваться как одна из важных характеристик. Среди современных антигипертензивных препаратов норваск обладает уникальными свойствами, способствующими значимому снижению АД, в основном за счет уменьшения жесткости сосудистой стенки крупных артериальных сосудов.
ЛИТЕРАТУРА
1. Agabiti-Rosei E., Mancia G., OґRourke M.F. et al. // Hypertension. – 2007.– Vol.50.– P.154–160.
2. Anonymous // Lancet. – 1977.– Vol.1.– P.1088–1099.
3. Asmar R.G., London G.M., OґRourke et al. // Hypertension.– 2001.– Vol.38.– P.922–926.
4. Boutouyrie P., Bussy C., Hayoz D. et al. // Circulation. – 2000.– Vol.101.– P.2601–2606.
5. Dahlof B., Sever S., Poulter N.R. et al. // Lancet.– 2005.– Vol.366.– P.895–906.
6. Delsing D.J., Jukema J.W., van de Wiel M.A. et al. // J. Cardiovasc. Pharmacol. – 2003.– Vol.42.–P.63–70.
7. Dhakam Z., Yasmin, McEniery C.M. et al. // J. Hypertens. – 2008.– Vol.26.– P.351–356.
8. Dzau V.J., Gibbon G.N. // Hypertension.– 1991.– Vol.18, suppl. III.– P.III115–III121.
9. Franclin S.S., Izzo J.I. In: Hypertension Primer. – 3rd ed. – Philadelphia, Lipincott Williams&Wilkins, 2003.– P. 170–175.
10. Franklin S.S., Gustin W.G., Wong N.D. et al. // Circulation. – 1997.– Vol.96.– P.308–315.
11. Franklin S.S., Larson M.G., Khan S.A. et al. // Circulation. – 2001.– Vol.103.– P.1245–1249.
12. Guidelines Committee. 2003 European Society of Hypertension-European Society of Cardiology guidelines for the management of arterial hypertension // J. Hypertens. – 2003.– Vol.21.– P.1011–1053.
13. 2007 Guidelines for the Management of Arterial Hypertension. The Task Force for the management of Arterial Hypertension of the European Society of Hypertension and of the European Society of Cardiology (ESC) // J. Hypertension. – 2007.– Vol.25.– P.1105–1187.
14. Hansen T., Staessen J., Pedersen B. et al. // Circulation. – 2006.– Vol.113.– P.664–670.
15. Heijden-Spek J.J., Staessen J.A., Fagard R.H. et al. // Hypertension. – 2000.– Vol.35.– P.637–642.
16. Izzo J.L. // J. Am. Geriartric Soc. – 1981.– Vol.29.– P.520–534.
17. Jondeau G., Boutouyrie P., Lacolley P. et al. // Circulation. – 1999.– Vol.99.– P.2677–2681.
18. Kelly R., Daley J., Avolio A. et al. // Hyper-tension. – 1989.– Vol.14.– P.14–21.
19. Kingwell B.A., Gatzka C.D. // Am. J. Hypertension.– 2002.– Vol.20.– P.2337–2340.
20. Laurent S., Cockcroft J., Van Bortel L. et al. // Eur. Heart J. – 2006.– Vol.27.– P.2588–2605.
21. Mahmud A., Feely J. // Expert. Rev. Cardiovasc. Ther.– 2003. Vol.1.– P.65–78.
22. Mancia G., Laurent S., Agabiti-Rosei E. et al. // J. Hypertens. – 2009.– Vol.27.– P.2121–2158.
23. Mancini G.B., Miller M.E., Evans G.W. et al. // Am. J. Cardiol. – 2002.– Vol.89.– P.1414–1416.
24. Mason R.P. // Atherosclerosis.– 2002.– Vol.165.– P.191–199.
25. Mattace-Raso F.U., van der Cammen T.J., Hofman A. et al. // Circulation. – 2006.– Vol.113.– P.627–663.
26. McEniery C.M., Wilkinson I.B., Avolio A.P. // Clinical and Experimental Pharmacology and Physiology.– 2007.– Vol.34.– P.665–671.
27. McEniery C.M., Yasmin, Hall I.R. et al. // J. Am. Coll. Cardiol.– 2005.– Vol.46.– P.1753–1760.
28. Mitchell G.F., Vita J.A., Larson M.G. et al. // Circulation. – 2005.– Vol.112.– P.3722–3728.
29. Mitchell G.F., Hwang S.-J., Vasan R.S. et al. // Circulation.– 2010.– Vol.121.– P.505–511.
30. Morgan T., Lauri J., Bertran D. et al. // Am. J. Hypertens.– 2004.– Vol.17.– P.118–123.
31. Najjar S.S., Scuteri A., Lakata E.G. et al. // Hypertension. – 2005.– Vol.46.– P.454–462.
32. Nichols W.W., OґRourke M.F. McDonaldґs blood flow in arteries.– 4th ed. – London: Amold, 1998.– 564 p.
33. OґRourke M.F. Arterial function in health and disease. – Edinburgh: Churchill, 1982. – 288 p.
34. OґRourke M.F., Safar M.E., Dzau V. (eds). Arterial Vasodilation: Mechanisms and Therapy. – London: Edward Arnold, 1993.
35. Poulter N., Sever P. Anglo-Scandinavian Cardiac Outcomes Trial. History, results and implications for the management of high blood pressure.– Birminham: Sherborne Gibbs Ltd, 2005. – 144 p.
36. Roman M.J., Devereux R.B., Kizer J.R. et al. // Hypertension. – 2007.– Vol.50.– P.197–203.
37. Roman M.J., Ganau A., Saba P.S. et al. // Hypertension. – 2000.– Vol.36.– P.489–494.
38. Roman M.J., Okin P.M., Kizer J.R. et al. // J. Hypertension. – 2010.– Vol.28.– P.384–388.
39. Safar M.E., Blasher J., Pannier B. et al. // Hypertension.– 2002.– Vol.39.– P.735–738.
40. Safar M.R., Levy B.I., Struijker-Boudier H. // Circulation. – 2003.– Vol.107.– P.2864–2869.
41. Schillaci G., Grassi G. // J. Hypertens. – 2010.– Vol.26.– P.237–239.
42. Schillaci G., Verdecchia P., Porcellati C. et al. // Hypertension. – 2000.–Vol.35.– P.580–586.
43. Schmitt M., Avolio A., Quasem A. et al. // Hypertension. – 2005.– Vol.46.– P.227–231.
44. Sever P.S., Dahlof B., Poulter N.R. et al. // J. Hypertension. – 2001.– Vol.19.– P.1139–1147.
45. Sever P.S., Dahlцf B., Poulter N.R. et al. // Lancet. – 2003.– Vol.361.– P.1149–1158.
46. Ting C.-T., Chen C.-H., Chang M.-S. et al. // Hypertension. – 1995.– Vol.26.– P.524–530.
47. Tsuchikura S., Shoji T., Kimoto E. et al. // J. Atheroscler. Thromb. – 2010.– Vol.17.– P.658–665.
48. Turnbull F. // Lancet.– 2003.– Vol.362.– P.1527–1535.
49. Weber T., Auer J., O’Rourke M.F. et al. // Circulation. – 2004.– Vol.109.– P.184–189.
50. Willams B., Lacy P.S., Thom S.M. et al. // Circulation.– 2006.– Vol.113.– P.1213–1225.
51. Williams B. // J. Hypertens. – 2007.– Vol.25.– P.1351–1353.
52. Williams B., Lacy P.S. et al. // J. Hypertens. – 2005.– Vol.23.– P.487–488.
53. Williams B., Lacy P.S. For the CAFЙ and the ASCOT (Anglo-Scandinavian Cardiac Outcomes Trial) Investigators.// J. Am. Coll. Cardiol. – 2009.– Vol.54.– P.705–713.
54. Wright J.T.J.R., Barkis G., Green T. et al. // JAMA.– 2002.– Vol.288.– P.2421–2431.
Медицинские новости. – 2010. – №10. – С. 24-30.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.