Остеопороз (ОП) — системное заболевание, для которого характерно снижение костной массы, нарушение микроархитектоники костной ткани, что приводит к слабости костей и увеличению риска переломов. Как показали эпидемиологические исследования, нет ни одной нации или страны, свободной от остеопороза. По данным американских исследователей, около 10 млн жителей США страдают ОП, из них около 80% женщины, и приблизительно у 34 млн американцев выявлено снижение костной массы, что является риском развития остеопороза. Ежегодно в США отмечается приблизительно 1,5 млн переломов из-за ОП, наиболее типичные места переломов — бедро, позвоночник и запястье. По сообщениям экспертов только в 2001 г. прямые затраты на лечение остеопоротических переломов составили около 17 млрд долларов США [12]. Ежегодно 1 из 8 жителей Европы старше 50 лет имеет перелом позвоночника. Согласно прогнозу экспертов 1 из 3 женщин и 1 из 9 мужчин старше 80 лет будут иметь перелом бедра в результате ОП. Старение населения приводит к увеличению числа остеопоротических переломов. Оценка мировой тенденции показала, что постарение популяции приведет к двукратному увеличению частоты переломов бедра между 2005—2050 гг. Последствия ОП в виде переломов позвоночника и трубчатых костей обусловливают значительный подъем заболеваемости, смертности и инвалидности населения.
Точная патофизиология остеопороза неизвестна; заболевание развивается при нарушении баланса образования и резорбции кости остеобластами и остеокластами соответственно. Цель лечения при остеопорозе — восстановление указанного равновесия путем подавления костной резорбции или стимуляции костеобразования [5]. Патогенетическая терапия ОП традиционно включает препараты, замедляющие костную резорбцию (бисфосфонаты, селективные модуляторы эстрогеновых рецепторов — СМЭР, эстрогены, кальцитонин); медикаменты, стимулирующие костеобразование (паратиреоидный гормон, фториды, анаболические стероиды, андрогены, соли стронция, гормоны роста); препараты многопланового действия (витамин Д, статины, оссеин-гидроксиапатитный комплекс). Многие из этих препаратов предотвращают будущую потерю костной массы, улучшают минеральную плотность костной ткани (МПКТ) и снижают риск переломов, однако при этом не происходит значительного усиления костеобразования.
В последнее время появились данные о тесной связи сосудистых заболеваний c патологией костной системы [15]. Хотя параллелизм указанных явлений нередко объясняют процессами старения, такая зависимость остается статистически значимой при скорректированном по возрасту анализе данных [8]. У женщин с ОП в постменопаузе отмечают достоверно более высокий риск патологии сердечно-сосудистой системы по сравнению с лицами такого же возраста без снижения минеральной плотности скелета [23]. Кроме того, у лиц с низкой минеральной плотностью костной ткани наблюдаются гиперлипидемия и более тяжелое течение атеросклероза венечных артерий, для них характерен более высокий риск смерти от сосудистых явлений [1,10, 15, 21]. По данным T. Yamaguchi et al. [26], у женщин в постменопаузе уровень холестерина липопротеинов высокой и низкой плотности соответственно прямо и обратно коррелирует с показателями минеральной плотности скелета как поясничных позвонков, так и дистальной части лучевой кости.
Установлено, что костная и сосудистая ткани имеют ряд общих морфологических и молекулярных свойств, а кальцификат сосудов состоит из тех же компонентов, что и костная ткань. Предполагается определенное сходство механизмов развития ОП и атеросклероза, наличие локальных тканевых факторов в регуляции процессов биоминерализации [15]. Костная ткань и костный мозг содержат эндотелиальные клетки, преостеобласты и остеокласты — производные моноцитов, причем все они являются также нормальными компонентами клеточных популяций сосудистой стенки. Как костная ткань, так и стенка артериальных сосудов в условиях атеросклеротического процесса содержат остеопонтин, остеонектин, остеокальцин, морфогенетический костный протеин (bone morphogenetic protein), матриксный Gla-протеин, коллаген I, оксид азота, а также матриксные везикулы. В патогенезе атеросклероза и остеопороза задействованы моноциты с дифференциацией в макрофаги с пенистой цитоплазмой в пределах сосудистой стенки и в остеокласты — в костной ткани. Каждый остеон (морфологическая единица кости) содержит центральный сосуд, выстланный изнутри эндотелием, локализованным на субэндотелиальном матриксе. Клетки-предшественники остеобластов расположены непосредственно по периферии матрикса. В сосудистой стенке находятся клеточные элементы, дифференцирующиеся в остеобласты в соответствии со стадиями дифференциации костных остеобластов, в итоге продуцирующих минеральный компонент кости [15]. Принципиально значимым является тот факт, что одни и те же оксидированные липиды, которые инициируют атеросклероз, индуцируют также минерализацию и дифференциацию остеобластов в стенке артерий [16].
В конце 90-х гг. ХХ в. стало известно, что синтез холестерина и активация остеокластов происходят при участии единого каскада биохимических процессов [11,17].
Синтез холестерина происходит в несколько этапов; 3-гидрокси-3-метилглутарил коэнзим А (ГМК-КоА) под влиянием редуктазы ГМК-КоА (подавляемый статинами энзим) превращается в мевалонат. Последний трансформируется в геранилпирофосфат, который в свою очередь превращается в фарнезилпирофосфат при участии фарнезилпирофосфатсинтетазы; активность ее ограничивается бисфосфонатами. В завершение образуются сквален и, наконец, холестерин [5]. Активация остеокластов происходит при участии промежуточного продукта описанного выше каскада — фарнезилпирофосфата, а также геранилгеранилпирофосфата (образуется из фарнезилпирофосфата). Процесс активации протекает путем модификации структуры триггерных внутриклеточных протеинов — глутамилтранспептидаз и ГТФаз — в ходе процесса пренилации [27].
Согласно данным S.P. Luckman et al. [17], а также E. van Beek et al. [22] образование активирующих остеокласты продуктов можно в равной мере эффективно ограничивать invitroс помощью бисфосфонатов и статинов, замедляющих продукцию мевалоната. Фактически при воздействии статинов или бисфосфонатов происходит гибель остеокластов посредством апоптоза. В результате снижается интенсивность резорбции кости и восстанавливается баланс костеобразования.
Другой механизм, посредством которого статины способны оказывать влияние на минеральный состав скелета, был открыт in vitro в 1999 г. G. Mundy et al. [13]. Речь идет об активации промотора морфогенетического костного протеина-2 (bone morphogenetic protein-2 — ВМР-2) — фактора роста, обусловливающего пролиферацию и созревание остеобластов, тогда как трансформирующий фактор роста и фактор роста фибробластов стимулируют пролиферацию остеобласта, но замедляют его дифференцировку [18].
Было показано, что ловастатин, а также другие липофильные статины — симвастатин, флувастатин и мевастатин усиливают экспрессию мРНК ВМР-2, в результате чего происходит более чем двукратное увеличение продукции указанного ростового фактора в клеточной культуре остеобластов [13]. Анаболические эффекты симвастатина были подтверждены в эксперименте на мышах, когда после введения 1 мкмоль симвастатина в течение 7 дней было выявлено увеличение количества остеобластов и объема остеоида на 30-50% [13].
В то же времяinvitro показано, что водорастворимый правастатин не влияет на BMP-2 [20].
Результаты дальнейших исследований свидетельствуют об увеличении объема трабекулярной костной ткани с 39 до 94% в условиях экспериментального постменопаузального остеопороза (самки крыс после овариэктомии) при пероральном введении симвастатина [5].
В клинических исследованиях получены данные о повышении минеральной плотности костной ткани в результате приема статинов больными сахарным диабетом [4], а также женщинами в постменопаузе [6].
После сообщения G. Mundy в 1999 г. на ежегодной конференции ASBMR были доложены результаты первых клинических исследований, посвященных влиянию статинов на костную ткань у людей. Проведено два метаанализа. В более раннем D.C. Bauer et al. [2] использовали данные четырех больших проспективных исследований (Study of Osteoporotic Fractures, Fracture Intervention Trial, Heart and Estrogen/Progestin Replacement Study, Rotterdam Study). Оценивались также данные обсервационных (8) и клинических (2) исследований. Была определена тенденция к меньшему числу переломов в группе лиц, принимавших статины. В метаанализе С. Hatzigeorgiou et al. [7] была проведена оценка 31 работы, включая 7 рандомизированных контролируемых исследований. В целом было отмечено протективное влияние статинов только на МПКТ и риск перелома в области проксимального отдела бедренной кости: риск перелома — OR 0,60 (95% CI 0.45, 0.78), а повышение МПКТ (Z score) — 0,12 (95% CI 0.05, 0.19). Действие статинов на переломы и МПКТ позвонков было незначительным. При анализе результатов, полученных только среди женщин, также определялось влияние статинов на область проксимального отдела бедра: риск перелома — OR 0,75 (95% CI 0.60, 0.95), повышение МПКТ (Z score) – 0,11 (95% CI 0.04, 0.18). Статины оказывали весьма незначительное влияние на маркеры костного обмена. Отмечалось снижение уровня щелочной фосфaтазы, повышение содержания N-терминальных телопептидов коллагена I типа, связанных поперечными сшивками; влияния на остеокальцин и C-терминальные телопептиды коллагена I типа не обнаружено.
В 3 проспективных исследованиях при использовании статинов у пожилых женщин отмечалась более низкая частота переломов бедра и других локализаций, кроме позвонков, но различия не были статистически достоверными [24]. В данных исследованиях отмечался незначительный прирост костной плотности. В исследовании типа случай — контроль при применении статинов у пациентов New Jersey Medicaid на 50% был ниже уровень переломов бедра (RR = 0.50, CI: 0.33, 0.76) [24]. При этом у лиц, принимающих статины, был выше вес, который оказывает протективный эффект в отношении риска переломов. Похожие данные получили Chan et al. [3] при исследовании у 928 женщин старше 60 лет: уровень переломов был ниже в группе лиц, получавших статины в течение 1 года (OR = 0,48, CI: 0.27, 0.83). Опубликованные в 2005 г. данные R.E. Scranton et al. [19] указывают на достоверное снижение частоты переломов (на 36%) у мужчин пожилого возраста, принимавших статины.
Таким образом, кардиоваскулярная патология и снижение минеральной плотности скелета тесно взаимосвязаны. Остеопения и остеопороз прогрессируют параллельно с атеросклеротическими поражениями. Продукты окисления липидов, способствующие прогрессированию атеросклероза, подавляют дифференцировку остеобластов с одновременным ускорением созревания такого рода клеточных элементов в сосудистой стенке [14]. В настоящее время выявлены два механизма действия статинов на кость: стимуляция экспрессии костного морфогенетического белка-2, что приводит к усилению дифференцировки остеобластов; воздействие на мевалонат, вызывающее подавление активности остеокластов и их апоптоз.
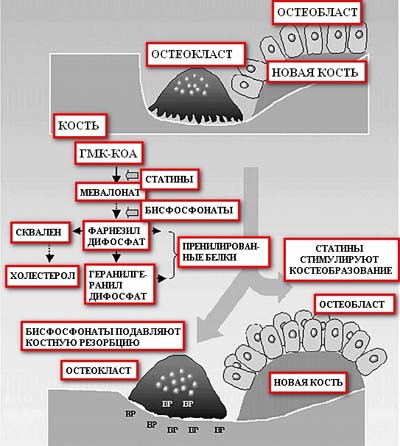
Рис. Ингибирование мевалоната и костный метаболизм. Примечание. ВР – бисфосфонаты.
Л и т е р а т у р а
1. Barengolts E.I., Berman M., Kukreja S.C. et al. // Calcif. Tissue Int. — 1998. — Vol. 62(3). — P. 209—213.
2. Bauer D.C., Mundy G.R., Jamal S.A. et al. // Arch. Intern. Med. — 2004. — Vol. 164(2). — P. 146—152.
3. Chan K.A., Andrade S.E., Boles M. et al. // Lancet. — 2000. — Vol. 355. — P. 2185—2188.
4. Chung Y.S., Lee M.D., Lee S.K. et al. // J. Clin. Endocrinol. Metab. — 2000. — Vol. 85(3). — P. 1137—1142.
5. Cruz A.С., Gruber B.L. // Clin. J. Med. — 2002. — Vol. 69(4). — P. 277—278, 280—282; 287—288.
6. Edwards C.J., Hart D.J., Spector T.D. // Lancet. – 2000. — Vol. 355(9222). — P. 2218—2219.
7. Hatzigeorgiou C., Jackson J.L. // Оsteoporosis Int. — 2005. — Vol. 16(8). — P. 990—998.
8. Jie K.G., Bots M.L., Vermeer C. et al. // Calcif. Tissue Int. — 2000. — Vol. 59(5). — P. 352—356.
9. LaCroix A.Z., Cauley J., Jackson R. et al. // J. Bone Miner. Res. — 2000. — Vol.15 (Suppl. 1). — P. 1066.
10. Laroche M., Pouilles J.M., Ribot C. et al. // Clin. Rheumatol. — 1994. — Vol. 13(4). — P. 611—614.
11.Luckman S.P., Hughes D.E., Coxon F.P. et al. // J. Bone Miner. Res. — 1998. — Vol. 13(4). — P. 581—589.
12.Michael J. Gonyeau, Pharm.D. // Pharmacotherapy. — 2005. — Vol. 25(2). — P. 228—243.
13. Mundy G., Garrett R., Harris S. et al. // Science. — 1999. — Vol. 286(5446). — P. 1946—1949.
14. Parhami F. // Essent. Fatty Acids. — 2003. — Vol. 68(6). — P. 373—378.
15. Parhami F., Garfinkel A., Demer L.L. // Arterioscler. Thromb. Vasc. Biol. — 2000. — Vol. 20(11). — P. 2346—2348.
16.Parhami F., Morrow A.D., Balucan J. et al. // Arterioscler. Thromb. Vasc. Biol. — 1997. — Vol. 17(4). — P. 680—687.
17. Russell R.G., Rogers M.J., Frith J.C. et al. // J. Bone Miner. Res. — 1999. — Vol. 14(Suppl. 2). — P. 53—65.
18. Sakou T. // Bone. —1998. — Vol. 22. — P. 591—603.
19. Scranton R.E., Young M., Lawler E. et al. // Arch. Intern. Med. — 2005. — Vol. 165(17). — P. 2007—2012.
20.Sugiyama M., Kodama T., Konishi K. et al. // Biochem. Biophys. Res. Commun. — 2000. — Vol. 271. — P. 688—692.
21. Uyama O., Yoshimoto Y., Yamamoto Y., Kawai A. // Stroke. — 1997. — Vol. 28(9). — P. 1730 —1732.
22. van Beek E., Pieterman E., Cohen L. et al. // Biochem. Biophys. Res. Commun. — 1999. — Vol. 264(1). — P. 108—111.
23. von der Recke P., Hansen M.A., Hassager C. // Am. J. Med. — 1999. — Vol. 106(3). — P. 273—278.
24. Wang P.S., Solomon D.H., Mogun H., Avorn J. // JAMA. — 2000. — Vol. 283. — P. 3211—3216.
25.Watanabe S., Fukumoto S., Takeuchi Y. et al. // Am. J. Med. — 2001. — Vol. 110(7). — P. 584—587.
26. Yamaguchi T., Sugimoto T., Yano S. et al. // Endocr. J. — 2002. — Vol. 49(2). — P. 211—217.
27.Zhang F.L., Casey P.J. // Ann. Rev. Biochem. — 1996. — Vol. 65. — P. 241—269.
Медицинские новости. – 2009. – №11. – С. 7-9.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.