Врожденная гиперплазия коры надпочечников (ВГКН) (адреногенитальный синдром, врожденная дисфункция коры надпочечников) объединяет группу моногенных заболеваний с аутосомно-рецессивным типом наследования, в основе которых лежат дефекты ферментов или транспортных белков, участвующих в процессах надпочечникового стероидогенеза [4, 6, 7, 10, 17]. Самая частая форма ВГКН – дефицит энзима 21-гидроксилазы (21-ГД), которая составляет от 90 до 95% всех вариантов адреногенитального синдрома [2, 7, 26]. По данным неонатальных скрининговых программ, проводимых в разных странах, популяционная частота встречаемости классических форм заболевания варьирует от 1:10 000 до 1:15 000 детей [2, 5, 13, 16].
Патофизиология дефицита 21-гидроксилазы
Фермент 21-ГД участвует в биосинтезе кортизола, превращая 17-гидроксипрогестерон (17-ОНП) в 11-дезоксикортизол [1, 4, 7]. Наблюдаемый при 21-гидроксилазной недостаточности дефицит кортизола приводит к повышению секреции адренокортикотропного гормона (АКТГ), вызывая компенсаторную гиперплазию коры надпочечников и последующую избыточную продукцию стероидов-предшественников ферментативного блока (17-ОНП) и андрогенов (андростендиона и тестостерона), синтез которых не зависит от процесса 21-гидроксилирования [7, 13]. Андростендион обладает менее выраженным андрогенным эффектом, однако способен метаболизироваться в тестостерон в периферических тканях. Гиперсекреция надпочечниковых андрогенов приводит к появлению клинических признаков ложного преждевременного полового развития у мальчиков и внутриутробной вирилизации у девочек [2, 17, 21].
Фермент 21-ГД участвует в синтезе минералокортикоидов. Гидроксилирование в положении 21 прогестерона приводит к образованию 11-дезоксикортикостерона, который является прогормоном альдостерона. По данным ряда исследований, до 70–75% детей с ВГКН наряду с глюкокортикоидной недостаточностью и андрогенизацией имеют минералокортикоидный дефицит различной степени выраженности [7, 29]. Он обусловлен снижением синтеза дезоксикортикостерона и альдостерона и проявляется ургентным состоянием уже в периоде новорожденности – развитием адреналового криза [1, 3, 17, 24, 29]. Между 4–15 днями жизни ребенка появляются неспецифические симптомы заболевания: вялое сосание, частые срыгивания, диарея, снижение массы тела. Характерно прогрессирующее нарастание электролитного дисбаланса, приводящее к развитию гипонатриемической дегидратации, метаболическому ацидозу и шоку. Эти лабораторные нарушения, являясь отражением многих состояний периода новорожденности (табл.1) [7], затрудняют диагностический поиск и часто приводят к ошибочному диагнозу у мальчиков с классической сольтеряющей формой ВГКН [19, 20]. Без адекватной заместительной рано начатой гормональной терапии дети погибают при явлениях острой надпочечниковой недостаточности [2, 5–7].
Таблица 1. Причины сольтеряющего синдрома в периоде новорожденности [7]
|
Растроинтестинальные потери
|
Патология надпочечников
|
Гастроэнтерит
Пилоростеноз
|
Врожденная гипоплазия надпочечников
Врожденная гиперплазия надпочечников
Врожденный гипоальдостеронизм
Псевдогипоальдостеронизм
|
|
|
Недоношенный новорожденный
Острый пиелонефрит
Почечная дисплазия
|
Классификация ВГКН
В зависимости от степени выраженности минералокортикоидной недостаточности, сроков появления андрогенизации выделяют классическую (сольтеряющую и простую вирильную) и неклассическую (позднюю) формы дефицита 21-ГД [6, 7, 17, 25, 29].
Различные варианты гормонального дисбаланса ВГКН у детей проявляются своим спектром клинических симптомов (табл. 2) [29].
Таблица 2. Клинические формы ВДКН вследствие дефицита 21-ГД [29]
|
Форма
|
Классическая сольтеряющая
|
Классическая простая вирильная
|
Неклассическая
|
|
Мальчик
|
Девочка
|
Мальчик
|
Девочка
|
Мальчик
|
Девочка
|
Возраст постановки диагноза
|
|
|
|
|
Постнатальный период, без возрастной очерченности
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
45–600 нмоль/л (АКТГ – стимулированный)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Глюко- и минералокортикоиды
|
|
Глюкокортикоиды (симптоматически)
|
Классическая форма ВГКН
Классическая форма 21-гидроксилазной недостаточности приводит к выраженной гиперандрогении. Новорожденные девочки с дефицитом 21-ГД имеют различную степень вирилизации наружных гениталий, обусловленную высоким внутриутробным уровнем надпочечниковых андрогенов, начиная с 7-й недели гестации [29]. При рождении отмечается гипертрофия клитора с формированием его головки, сращение мошоночного шва, формирование урогенитального синуса и отсутствие пальпируемых гонад [29]. В редких случаях внутриутробная андрогенизация выражена настолько, что наружные половые органы пациентки практически соответствуют мужским [2, 5, 7, 29]. При ультразвуковом исследовании органов малого таза визуализируются внутренние гениталии (матка, фаллопиевы трубы и яичники).
Все новорожденные с фенотипическим мужским полом и не пальпируемыми в мошонке и по ходу паховых каналов яичками, а также дети с бисексуальным строением гениталий должны быть обследованы для исключения ложного женского гермафродитизма с обязательным кариотипированием [8, 9, 13, 15, 17]. Отсутствие сонографического определения матки у новорожденной девочки не является достоверным диагностическим признаком и может наблюдаться при загибе матки кзади [7].
Степень выраженности вирилизации девочек с дефицитом 21-ГД обусловлена типом мутации структурных генов фермента, индивидуальными различиями активности андрогеновых рецепторов и особенностями метаболизма стероидов-предшественников андрогенов [14].
Строение наружных половых органов новорожденных мальчиков с 21-гидроксилазной недостаточностью соответствует генетическому полу. Они имеют нормально сформированный половой член, мошонку, яички и внутренние вольфовы протоки [29]. Может наблюдаться увеличение полового члена, вторичная пигментация мошонки и сосков вследствие повышенной секреции меланоцитостимулирующего гормона [7].
В постнатальном периоде при позднем установлении диагноза и отсутствии адекватного лечения детей с 21-гидроксилазным дефицитом отмечаются ускорение темпов физического развития, преждевременная костная дифференцировка с ранним закрытием эпифизарных зон, формирование маскулинного телосложения, увеличение клитора или полового члена, преждевременное появление полового оволосения [5, 6, 29]. Окончательный рост взрослых больных с адреногенитальным синдромом ниже генетического на 1–2 стандартных отклонения [11]. Истинный пубертат у нелеченых пациентов обоего пола наступает поздно. У девочек нарушения со стороны репродуктивной системы проявляются в виде олигоменореи, аменореи, меноррагий, бесплодия [6, 7], для мальчиков характерными признаками являются гипоплазия яичек и олиго/азооспермия [6, 7, 29].
Лабораторное подтверждение классической формы дефицита 21-ГД у новорожденных основано на определении значительно повышенных уровней 17-ОНП, который является патогенетическим маркером заболевания (табл. 2) [2, 7, 29]. Базальный уровень этого гормона (более 300 нмоль/л) примерно в 100 раз превышает нормальные показатели (менее 3 нмоль/л) для гестационного возраста и массы тела ребенка [5, 8, 13, 29].
У недоношенных, детей с родовыми травмами или рожденных с низким весом при нормальных сроках гестации концентрации 17-ОНП могут быть повышенными и при отсутствии дефицита этого фермента [8]. В этих случаях рекомендуется повторное определение гормонального показателя с интервалом 5–7 дней. Снижение уровней 17-ОНП в динамике позволяет исключить 21-гидроксилазную недостаточность [17].
Для своевременного выявления гормональных нарушений, характерных для дефицита 21-ГД, необходимо обязательно обследовать всех новорожденных с гермафродитными наружными половыми органами и мальчиков с синдромом потери соли [8, 9, 17]. Необходимо помнить, что одной из самых распространенных причин бисексуального строения гениталий у детей является ВГКН, обусловленная недостаточностью 21-ГД [8].
Кариотип 46ХХ при гермафродитных наружных половых органах, повышенный уровень 17-ОНП для данных сроков гестации и массы тела ребенка, наличие матки при ультразвуковом исследовании с 95% вероятностью свидетельствуют о 21-гидроксилазной недостаточности у девочек [8, 9].
У мальчиков с классической формой дефицита 21-ГД отправными точками диагностического поиска являются отсутствие адекватного увеличения массы тела, наличие клинических и лабораторных признаков (гипонатриемия, гиперкалиемия, гиперренинемия, метаболический ацидоз) сольтеряющего компонента [29]. Простая вирильная форма заболевания у них диагностируется только к 4–5 годам при манифестации признаков ложного преждевременного полового развития по изосексуальному типу.
Классическая сольтеряющая форма ВГКН не проявляется у новорожденных и в первые дни жизни ребенка. Дифференциальный диагноз с простой вирильной формой заболевания проводится на основании мониторирования сывороточных или плазменных уровней электролитов, активности ренина плазмы, проведения стимуляционной пробы с АКТГ, результатов молекулярного анализа генов 21-ГД [7–9, 17, 29].
Неклассическая форма ВГКН
Неклассическая форма ВГКН представляет собой «мягкий» вариант проявления 21-гидроксилазного дефицита [7, 29] и является одним из наиболее распространенных аутосомно-рецессивных нарушений в ряде этнических групп [4]. Высокая степень встречаемости поздней формы адреногенитального синдрома отмечена у евреев ашкенази (1:27), в испанской (1:53), югославской (1:63), итальянской (1:333) популяциях и (1:1000) у других представителей европеоидной расы, 40% которых – англосаксонского происхождения [23].
Особенностью манифестации этого варианта заболевания является постнатальная андрогенизация разной степени выраженности без четкой очерченности возрастного периода [5, 7, 18, 29]. Для детей обоего пола допубертатного возраста характерно небольшое ускорение роста, опережение костного возраста, преждевременное адренархе (пубархе) и акне [29]. У девочек может наблюдаться гипертрофия клитора и высокая задняя спайка промежности [29], у мальчиков – рост полового члена без увеличения объема яичек [18, 19, 29]. В пубертатном и постпубертатном возрасте андрогенизация клинически проявляется гирсутизмом, нарушениями функции репродуктивной системы (синдромом поликистозных яичников, нерегулярным менструальным циклом, бесплодием и невынашиванием беременности) [7, 18]. Многие пациенты имеют бессимптомное течение заболевания [29].
Гормональные критерии диагностики ферментативных нарушений при неклассической форме ВГКН предложены M. New et al. [6]. Не установлено достоверных различий в уровнях плазменного тестостерона и дегидроэпиандростендиона у пациентов с неклассической формой ВГКН и у больных с овариальными формами истинной гиперандрогении [7]. Более того, девочки с поздним проявлением 21-гидроксилазного дефицита часто могут иметь нормальные уровни дегидроэпиандростендиона [7]. Дифференциальная диагностика форм гиперандрогении основана на выявлении повышенных значений 17-OHП при проведении пробы с АКТГ [7, 29]. У девушек и молодых женщин проба выполняется в фолликулярную фазу менструального цикла. Для поздних нарушений надпочечникового стероидогенеза характерно увеличение базального и стимулированного уровней 17-OHП более чем на 2 сигмальных отклонения от нормальных показателей. Пациенты, у которых только стимулированные уровни 17-OHП более чем на 2 сигмы превышают показатели контроля, но ниже значений 17-OHП при неклассическом варианте заболевания, относятся к категории гетерозиготного носительства [29].
Молекулярная диагностика ВГКН
Молекулярная диагностика дефицита 21-ГД основана на выявлении мутаций как следствие рекомбинации между двумя структурными генами фермента [4]. Оба гена локализованы на коротком плече 6-й хромосомы рядом с главным комплексом гистосовместимости (HLA – антигеном лейкоцитов человека). Один ген кодирует гидроксилазу-В (21-ГД-В), второй (псевдоген) – гидроксилазу А (21–ГД-А). 98% нуклеотидной последовательности генов 21-ГД-В и 21–ГД-А идентичны [4, 8]. Псевдоген содержит ряд мутаций, который делает невозможным процесс считывания информации на матричную РНК и, таким образом, лишает его активности. Особенности структуры генов 21-ГД создают предпосылки для многочисленных вариантов мутаций.
В настоящее время описано более 50 различных мутационных аллелей, приводящих к частичной или полной потере ферментативной активности 21-ГД [5, 7, 15, 29]. Активность мутантных форм 21-ГД, которые имеют специфические замены аминокислот и встречаются у гомозиготных и гетерозиготных больных с недостаточностью этого энзима, в основном коррелирует с клинической картиной заболевания (табл. 3) [4, 7, 8, 15].
Таблица 3. Взаимосвязь между клиническими формами дефицита 21-гидроксилазы и наиболее частыми генными мутациями (по C.L. Acerini, I.A. Hugnes, 1999)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Сольтеряющая форма обычно
|
|
|
|
|
|
|
|
|
Выраженная сольтеряющая форма недостаточности 21-ГД связана с тремя мутациями, которые полностью блокируют синтез нормального белка [4, 5]. К ним относятся делеция гена 21-ГД-В или замена части активного гена на псевдоген, образование стоп-кодона в экзоне 8 (глутамин 318 → стоп-кодон) [4] и делеция восьми пар нуклеотидов в экзоне 3. Последние две мутации составляют 4–7 и 3–10% всей молекулярной патологии ВГКН соответственно [4].
Наиболее часто встречается неделеционная мутация во 2-м интроне (26% всех мутаций), которая вызывает значительное снижение активности фермента 21-ГД [4]. Она выявляется как при тяжелой сольтеряющей, так и при простой вирильной форме ВГКН.
У больных с сольтеряющей формой заболевания установлены кластер мутации с заменой Ile–Val–Glu–Met (235 –238) на Asn–Glu–Glu–Lys [4] и одиночная точечная замена аргинина (356) на триптофан [5]. Часто регистрируются несколько мутаций в одной хромосоме [4].
Многочисленные точечные мутации структурных генов 21-ГД (в позиции Ile 172 Asn, Pro 30 Leu, Val 281 Leu и др.) клинически характеризуются различной степенью вирилизации пациента с развитием простого вирильного варианта заболевания или его поздней формы [8, 15].
Выявление конкретных мутаций гена помогает подтвердить диагноз и дифференцировать форму ВГКН, решить вопрос о необходимости начала терапии [2, 17, 29].
Проблемой, сдерживающей повсеместное введение молекулярной диагностики адреногенитального синдрома, в частности в качестве второго этапа неонатального скрининга, является относительная длительность проведения генетического анализа и его стоимость. Чувствительность генетического исследования недостаточно высока, учитывая возможность наличия неописанных мутаций [2] и отсутствие корреляции в малом проценте случаев между клиническими проявлениями заболевания и молекулярными дефектами [17].
Неонатальный скрининг ВГКН
В настоящее время программы неонатального скрининга на адреногенитальный синдром внедрены во многих странах, в том числе США, Франции, Германии, Италии, Швейцарии, Японии, Швеции и др. [2, 17, 29]. Массовое скрининговое обследование новорожденных на выявление 21-гидроксилазного дефицита рекомендовано принятым в 2002 г. консенсусом Европейского общества детских эндокринологов (ESPE) [17]. При анализе распространенности ВГКН по данным только клинической диагностики или при проведении скрининга результативность последнего подтверждается увеличением почти в 2 раза частоты заболевания и повышением соотношения мужчин и женщин до единицы [2].
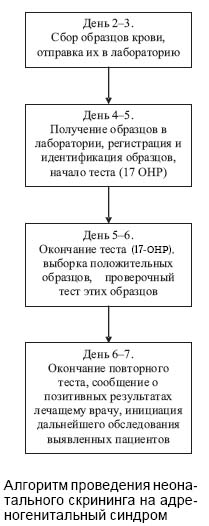
Эффективность процедуры неонатального скрининга на ВГКН определяется [2]:
• ранним, быстрым и полным сбором образцов крови у новорожденных (на 2–3-е сутки жизни);
• быстрым, надежным и относительно дешевым методом определения 17-ОНП;
• оптимальным выбором критериев диагностики заболевания (исключение ложноотрицательных и минимизация ложноположительных проб);
• наличием мобильной и своевременной системы передачи информации о положительных случаях ВГКН;
• ранним началом заместительной гормональной терапии.
Примерный алгоритм проведения скрининга новорожденных на адреногенитальный синдром представлен на рисунке [2, 17].
Эффективность неонатального скрининга на ВГКН заключается в возможности более раннего выявления больных обоего пола, правильного выбора половой принадлежности и регистрации паспортного пола, предотвращения развития острого надпочечникового криза, кардиогенного шока, его последствий и смерти ребенка [12, 22, 27, 30].
В заключение следует подчеркнуть, что ВГКН, обусловленная дефицитом 21-ГД, продолжает широко исследоваться с точки зрения как клинических и гормональных проявлений, так и молекулярной генетики. К настоящему времени определены молекулярные механизмы различных форм этого заболевания. Практическим результатом генетических разработок стало повышение точности пренатальной и ранней постнатальной диагностики, установление взаимосвязи между клиническими проявлениями адреногенитального синдрома и вызывающими их гормональными и молекулярно-генетическими нарушениями. Актуальное направление детской эндокринологии – совершенствование методики проведения массового неонатального скрининга данного заболевания. Улучшение и расширение возможностей диагностики ВГКН позволит оптимизировать протоколы лечения и наблюдения больных с адреногенитальным синдромом с учетом пола, возраста, клинической ситуации, поможет предотвратить прогрессирование патологического процесса и его осложнений, минимизировать побочные эффекты заместительной гормональной терапии.
1. Дедов И.И., Семичева Т.В., Петеркова В.А. // Половое развитие детей: норма и патология. – М.: Колор Ит Студио, 2002. – С. 119–129.
2. Кареева М.А., Семичева Т.В., Петеркова В.А. // Вопросы практ. педиатрии. – 2006. – № 4. – С. 102–104.
3. Клиническая фармакология / под ред. А.Г. Гилманна. – М.: Практика, 2006. – С. 1269–1280.
4. Молекулярная эндокринология / под ред. Б.Д. Вайнтрауба. – М.: Медицина, 2003. – С. 440–458.
5. Петеркова В.А., Семичева Т.В., Кузнецова Э.С. и др. // Врожденная дисфункция коры надпочечников у детей. – М., 2003. – С. 19–44.
6. Самсонова Л.Н., Зубкова Н.А. // Фарматека. – 2003. – № 8. – С. 63–65.
7. Acerini C.L., Hugnes I.A. // Topical endocrinology. – 1999. – N 13. – P. 14–18.
8. Bode H.H., Rivkees S.A., Cowley D.M. et al. // J. Pediatr. – 1999. – Vol. 134. – P. 185–189.
9. Charmandari E., Lichtarowicz-Krynska E.J., Hindmarsh P.C. // Arch. Dis. Child. – 2001. – Vol. 85. – P. 26–28.
10. Charmandari E., Brook C.G., Hindmarsh P.C. // Arch. Dis. Child. – 2002. – Vol. 86. – P. 266–269.
11. Eugster E.A., Dimeglio L.A., Wright J.C. et al. // J. Pediatr. – 2001. – Vol. 138. – P. 26–32.
12. Honour J.W., Torresani T. // Horm. Res. – 2001. – Vol. 55. – P. 206–211.
13. Hughes I.A. // Seminars in reproductive medicine. – 2002. – Vol. 20, N 3. – P. 229–241.
14. Iluizenda N.A., Koper J.W., De Lange P. et al. // Clin. Endocrinol. Metab. – 1998. – Vol. 83. – P. 144–151.
15. Jansen M., Wit J.M., Brande J.L. // Acta Paediatr. Scand. – 1981. – Vol. 70. – P. 229–233.
16. Jinno K., Sakura N., Nomura S. et al. // Paediatr. Intern. – 2001. – Vol. 43. – P. 478–482.
17. Joint LWPES/ESPE CAH working Group // Horm. Res. – 2002. – Vol. 58. – P. 188–195.
18. Kohn B., Levine L.S., Pollak M.S. et al. // Clin. Endocrinol. Metab. – 1998. – Vol. 83. – P. 144–151.
19. Moran C., Azziz R., Carmina E. et al. // Amer. J. Obstetr. Gynecol. – 2000. – Vol. 183. – P. 1468–1474.
20. Mullis P.E., Hindmarsch P.C., Brook C.G. // Eur. J. Pediatr. – 1990. – Vol. 150. – P. 22–25.
21. Nordenstrom A., Thilen A., Hagenfeldt L. et al. // Clin. Endocrinol. Metab. –1999. – Vol. 84. – P. 1505–1509.
22. Pass K.A., Lane P.A., Fernhoff P.M. et al. // J. Pediatr. – 2000. – Vol. 137. – P. 1–46.
23. Speiser P.W., Dupont B., Rubinstein P. et al. // Amer. J. Hum. Genet. – 1985. – Vol. 37. – P. 650.
24. Speiser P.W., Agdere L., Ueshiba H. et al. // New Engl. J. Med. – 1991. – Vol. 324. – P. 145–149.
25. Sperling M.A. // Pediatric Endocrinology. – New Jork: Alan R.Liss, 2002. – P. 385–438.
26. Therrelli B.L. // Endocrinol. Metab. Clin. North Amer. – 2001. – Vol. 30. – P. 15–30.
27. Tomlinson J.W., Stewart P.M. // Best Pract. Res. Clin. Endocrinol. Metab. – 2001. – Vol. 15. – P. 61–78.
28. Tomlinson J.W., Draper N., Mackie J. et al. // Clin. Endocrinol. Metab. – 2002. – Vol. 87. – P. 57–62.
29. White P.C., Speiser P.W. // Endocrine Reviews. – 2002. – Vol. 16, N 2. – P. 273–288.
30. Working Group on Neonatal Screening of the European Society for Pediatric Endocrinology: Procedure for neonatal screening for congenital adrenal hyperplasia due to 21-hydroxylase deficiency // Horm. Res. – 2001. – Vol. 55. – P. 201–205.
Медицинские новости. – 2009. – №2. – С. 6-9.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.