|

С.Л. Кабак, Ю.С. Кабак
Медиаторы локальной резорбции костной ткани при хроническом апикальном (верхушечном) периодонтите
Белорусский государственный медицинский университет
Апикальный (верхушечный) периодонтит — воспалительный процесс, который развивается вокруг верхушки корня зуба как осложнение кариеса и пульпита. Исходом хронического течения болезни является разрушение костной ткани. Очаг деструкции выявляется на рентгеновских дентальных снимках, и в периоды обострения периодонтита его размеры увеличиваются.
Еще в 1965 г. S. Kakehashi et al. в эксперименте показали, что микроорганизмы, инфицирующие пульпу корневых каналов, являются этиологическим фактором развития периапикального очага воспаления [25]. За счет выделения протеолитических ферментов они могут непосредственно вызывать повреждение периапикальных тканей [44]. Однако в настоящее время общепризнанной считается концепция, согласно которой развитие воспаления и последующее разрушение костной ткани вокруг верхушки корня зуба стимулируют не сами бактерии, а их антигены (схема), в том числе компоненты клеточной мембраны [81]. Одним из таких бактериальных остеолитических факторов является липополисахарид (эндотоксин), который представляет собой сложный гликолипид, состоящий из гидрофильной полисахаридной части и гидрофобного домена, известного под названием липид А и определяющего большинство биологических эффектов эндотоксина [29]. С помощью липополисахарида, выделенного из Fusobacterium nucleatum и Porphyromonas endodontalis —по статистике самых распространенных возбудителей эндодонтической патологии у человека, апикальный периодонтит воспроизводится в эксперименте [18]. Полимиксин (специфический ингибитор эндотоксина) подавляет развитие воспалительного процесса вокруг верхушки корня зуба у экспериментальных животных. Дополнительным этиологическим фактором развития периапикального воспаления является бактериальная ДНК с определенной последовательностью неметилированных CG динуклеотидов, которая стимулирует развитие иммунно-воспалительных реакций в тканях вокруг верхушки корня зуба [30, 33, 34].
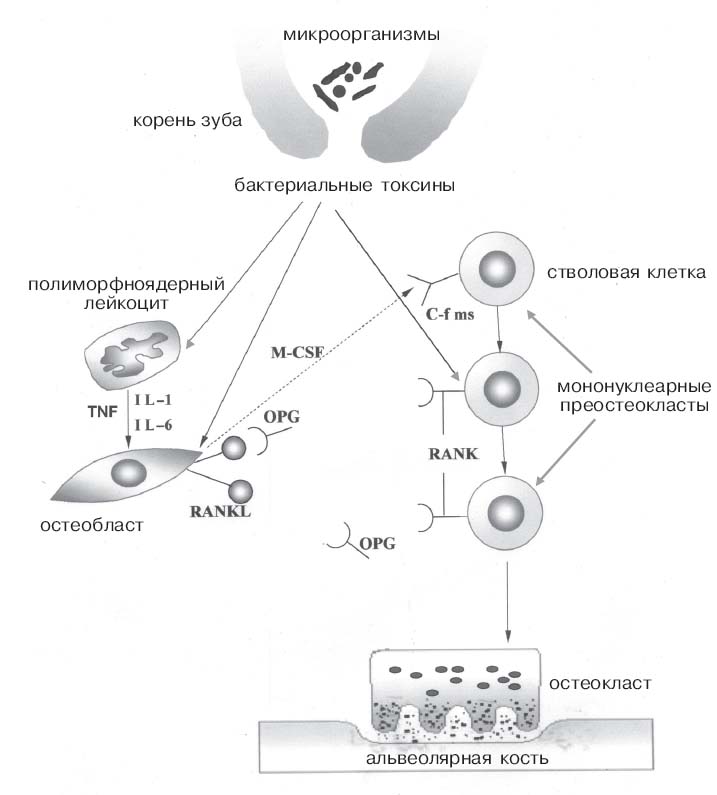
Схема. Медиаторы периапикальной деструкции костной ткани при инфицировании канала корня зуба и развитии апикального периодонтита
Бактериальные токсины непосредственно стимулируют продукцию транскрипционных факторов, предопределяющих дифференцировку остеокластов [44, 85]. В связи с этим им отводится ведущая роль в патогенезе локальной костной деструкции при хроническом апикальном периодонтите. Под действием LPS индуцируется экспрессия фактора некроза опухолей (TNF-α) и интерлейкина-1β (IL-1β) в клетках предшественниках остеокластов. В свою очередь эти цитокины являются мощным стимулятором дифференцировки остеокластов [87].
Кроме того, под действием липополисахаридов и других бактериальных антигенов в околоверхушечной области развивается комплекс иммуновоспалительных реакций [41, 64, 67]. При этом неспецифический иммунный ответ включает миграцию в очаг воспаления полиморфноядерных лейкоцитов и моноцитов/макрофагов. В результате взаимодействия Toll-like-рецепторов клеточной мембраны этих клеток с компонентами бактериальной стенки (LPS) экспрессируются цитокины, в том числе провоспалительные [19].
Локальные индукторы деструкции костной ткани [44]
· Простагландины
· Лейкотриены
· Провоспалительные цитокины (IL-1, IL-6, TNF)
· Факторы роста
· Бактериальные продукты:
- Липополисахариды (LPSs);
- Тейхоевые кислоты;
- Липид А-ассоциированные белки;
- Порины;
- cpn60 белки A. actinomy-cetemcomitans и E. coli;
- Гапстатин A. actinomy-cetemcomitans (8-kDa белок, обладающий антипролиферативным действием);
- Pasterella multocida toxin (PMT);
- дерматонекротический токсин (dermonecrotic toxin, DNT) B. bronchiseptica.
· Компоненты бактериальной стенки:
Мембранные белки A. actinomycetemcomitans, P. gingivalis, E. corrodens, Staphylococcus aureus, и Staphylococcus epidermidis;
Мембранные полисахариды A. actinomycetemcomitans;
32- kDa и 60-kDa мембранные белки Staphylococcus aureus;
43-kDa фимбриальный белок P. gingivalis
С помощью иммуногистохимических и радиоиммунологических методов в составе очага воспаления вокруг верхушки корня зуба и окружающей костной ткани выявляются клетки, продуцирующие цитокины — IL-1α, IL-2, IL-4, IL-5, IL-6, IL-10, IL-11, TNF-α [13, 28, 54, 63, 64, 70, 71, 80], фактор, стимулирующий образование колоний гранулоцитов/макрофагов (granulocyte-macrophage colony-stimulating factor, GM-CSF) [49], интерферон-γ [23, 80] и RANKL [52]. Используя метод полимеразной цепной реакции, L. Mei et al. в очаге периапикального воспаления, воспроизведенного у крысы, на 14-й день эксперимента (острая фаза воспалительного процесса) обнаружили пик экспрессии mRNA IL-1α, и TNF-α. Экспрессия mRNA IL-1β на те же сутки развития заболевания была выражена в меньшей степени [43].
В образцах периапикального экссудата, полученных у человека во время эндодонтического лечения апикального периодонтита, H. Shimauchi et al. выявили присутствие IL-18 и оксида азота (NO) [52]. Повышенное содержание IL-18 определялось у пациентов, у которых на момент забора материала регистрировались клинические симптомы хронического апикального периодонтита. В период обострения заболевания в периапикальном экссудате отмечается также увеличение концентрации простагландина Е2 (PGE2) [68]. Кроме того, авторы установили, что по мере увеличения размеров очага костной деструкции на рентгеновских снимках содержание PGE2 в периапикальном экссудате возрастает.
В периапикальном экссудате выявляются одновременно IL-1β и антагонист рецептора к IL-1β (IL-1 receptor antagonist, IL-1ra). При этом у пациентов в период обострения хронического апикального периодонтита количество IL-1β по сравнению с IL-1ra значительно выше, чем у пациентов без клинических симптомов болезни [55].
Очаги периапикального воспаления, содержащие эпителиальные клетки и сопровождающиеся клиническими симптомами хронического апикального периодонтита, T. Radics et al. характеризуют как иммунологически активную стадию развития болезни, для которой характерен высокий уровень содержания IL-6 и GM-CSF [49]. Вместе с тем отсутствие симптомов заболевания, эпителиальных клеток и низкое содержание IL-6 и GM-CSF свидетельствует, по мнению авторов, о прогрессировании репаративных процессов в области верхушки корня зуба.
Резорбцию кости как при ее физиологической перестройке, так и при патологии осуществляют остеокласты, которые так же, как моноциты и макрофаги являются производными гемопоэтической стволовой клетки [40, 42]. В процессе многоступенчатой дифференцировки мононуклеарные клетки-предшественники остеокластов сначала приобретают специфические маркеры фенотипа, такие как кальцитониновые и β3 интегриновые рецепторы, а также способность синтезировать тартрат-резистентную кислую фосфатазу (tartrate-resistant acid phosphatase, TRAP) и катепсин К. Затем мононуклеарные преостеокласты сливаются и формируют гигантские многоядерные клетки, способные осуществлять резорбцию костной ткани за счет широкого арсенала лизосомальных ферментов (металлопротеиназ и других протеаз). Один активированный остеокласт способен разрушить до 200 000 мм3 кости в день. Такое количество костной ткани продуцирует 7—10 генераций остеобластов, средняя продолжительность жизни которых составляет 15—20 дней [62].
В настоящее время главными регуляторами дифференцировки остеокластов считаются лиганд рецептора активатора ядерного фактора каппа Б — фактор, стимулирующий образование колоний макрофагов, а также остеопротегерин (рисунок).
Лиганд рецептора активатора ядерного фактора каппа Б(RANK-ligand, RANKL; TRANCE [tumor necrosis factor (TNF) related activation induced cytokine], OPGL [osteoprotegerin ligand]) является представителем семейства лигандов для рецепторов фактора некроза опухолей и продуцируется стромальными клетками костного мозга, остеобластами, а также лимфоцитами (активированными антиген-специфическими CD4 положительными Т-лимфоцитами). У человека клетки периодонтальной связки обладают фенотипом, сходным с остеобластами, и способны продуцировать лиганд рецептора ядерного фактора каппа Б [14]. Существенное увеличение содержания RANKL в жидкости из десневого кармана регистрируется при маргинальном периодонтите, сопровождающемся выраженной резорбцией альвеолярной кости [77]. Лиганд связан с плазмолеммой продуцирующих его клеток и взаимодействует с соответствующим рецептором (RANK (receptor activator of NF-Kappa B) — рецептор активатор ядерного транскрипционного фактора каппа Б) на поверхности клеток-предшественников остеокластов, что ведет к их дифференцировке [38, 74]. Существует несколько путей внутриклеточной передачи сигнала от связанных с лигандом рецепторов плазмолеммы, в которых задействованы разные вторичные посредники.
Взаимодействие RANKL с рецептором активирует факторы семейства TRAF (TNF receptor activating factor), в состав которого входят TRAF 2, 5 и 6 [82]. Установлено, что из этих трех факторов только TRAF 6 может самостоятельно индуцировать остеокластогенез [24]. Различные домены TRAF 6 модулируют начало дифференцировки клеток-предшественников остеокластов и их последующее созревание за счет активации разных киназных каскадов [31]. В частности, под его влиянием запускается каскад митогенактивированных протеинкиназ (mitogen-activated protein kinases, MAPK). Мишенью сигнала, передаваемого с помощью протеинкиназ, являются протоонкогены с-fos и с-jun, которые кодируют белки (c-Jun и c-Fos), являющиеся основными компонентами многосубъектного фактора транскрипции АР-1 (активирующеего протеина-1), предопределяющего рост, пролиферацию и дифференцировку клеток. F. Ikeda et al. считают, что RANKL-индуцированное образование остеокластов in vitro и in vivo осуществляется главным образом благодаря активации c-Jun сигнального пути [21].
Второй путь передачи сигнала — активация ядерного фактора транскрипции каппа Б (NFkB). Этот процесс также происходит в значительной степени за счет участия в качестве вторичного мессенджера факторов семейства TRAF (TRAF2), передающих управляющий сигнал через IKK — ингибитор kБ киназного комплекса (Ik) [74]. По действием IKK происходит фосфорилирование kБ киназного комплекса и его отсоединение от транскрипционного фактора NFkB. В результате транскрипционный фактор получает возможность транслоцироваться к ядру и активировать транскрипцию генов, определяющих дифференцировку остеокластов.
Имеются данные, что RANKL также регулирует уровень катепсина К — фермента, который продуцируется остеокластами и играет ключевую роль в резорбции костной ткани [10]. Вместе с тем RANKL индуцирует ген IFN-β в клетках-предшественниках остеокластов, в результате чего задерживается их дифференцировка [69]. В основе торможения остеокластогенеза под действием IFN-β лежит нарушение RANKL-индуцированной экспрессии транскрипционного фактора c-Fos.
Экспрессия RANKL клетками-мишенями активируется под воздействием фактора некроза опухолей (TNF-α) [17], простагландинов (PG2) [39], IL-1 и IL-11 [3, 17], липополисахаридов [29], бактериальной CpGp-DNA и двухнитевой вирусной ДНК [33, 88] и фактора роста фибробластов (FGF-2) [9].
Фактор, стимулирующий образование колоний макрофагов (macrophage colony-stimulating factor, M-CSF), продуцируется стромальными клетками костного мозга и остеобластами, связывается с рецепторами тирозинкиназы (c-fms) на поверхности клеток-предшественников остеокластов и делает их готовыми к дальнейшему развитию. При отсутствии фактора дифференцировка останавливается на стадии преостеокластов. У мутантных мышей с врожденным отсутствием фактора, стимулирующего образование колоний макрофагов, развивается остеопетроз (относительное увеличение содержания костной ткани в составе костей). Рядом исследований доказано, что только в присутствии M-CSF может реализовываться взаимодействие RANKL c соответствующим рецептором на плазмолемме клеток, которые должны дифференцироваться в остеокласты. Кроме того, увеличивается продолжительность существования этих клеток[37].
Дифференцировку остеокластов тормозит остеопротегерин (osteo-protegerin, OPG, osteoclasto-genesis-inhibitory factor, OCIF) — гликопротеин, который функционально представляет собой рецептор-ловушку (приманку) из семейства TNF-рецепторов, лишенный трансмембранного домена [50]. Установлено, что остеопротегерин вырабатывается остеобластами, а также фибробластами периодонтальной связки [14, 79], и блокирует взаимодействие RANK и RANKL, в результате чего невозможна активация клеток-предшественников остеокластов. Многие из тех факторов, которые стимулируют экспрессию RANKL (включая IL-1α, PGE2 и IL-11), подавляют продукцию OPG [3, 26, 51, 60, 78], что в еще большей степени стимулируют остеокластогенез и резорбцию костной ткани.
Цитокины IL-13, INF-γ и TGF-β1, наоборот, активируют продукцию OPG и/или вызывают супрессию RANKL [47]. В культуре тканей из опорных клеток и предшественников остеокластов M. Karst et al. при низкой концентрации TGF-β отмечали высокое значение соотношения RANKL/OPG [27]. По мере нарастания концентрации трансформирующего фактора роста β продукция OPG все больше преобладала над экспрессией RANKL. Одновременно авторы фиксировали супрессию фактора, стимулирующего образование колоний макрофагов (M-CSF).
Действие продуктов локального неспецифического иммунного ответа (IL-1 и TNF) на процесс костной деструкции в определенной степени осуществляется через RANKL/RANK/NFkB сигнальный путь регуляции остеокластогенеза [14, 48, 61]. Одновременно имеет место прямое воздействие цитокинов на остеокласты [7, 35, 65, 72]. На клеточной мембране этих клеток экспрессируется два типа IL-1-рецепторов [83].
Ключевую роль IL-1 и TNF в патогенезе деструкции костной ткани доказывают экспериментальные работы, в которых интенсивность деструкции модулируется блокированием биологического действия провоспалительных цитокинов. В максимальных концентрациях моноклональные антитела к TNF-α снижают остеокластогенную активность цитокина, индуцированную бактериальными продуктами, примерно на 40—60% [22]. На 60% уменьшается размер очага костной деструкции у животных с экспериментально воспроизведенным апикальным периодонтитом после введения антагониста рецептора IL-1 [63]. На экспериментальной модели ревматоидного артрита S.B. Abramson, A. Amin отмечали уменьшение интенсивности эрозии костной ткани после введения моноклональных антител к IL-1 и антагониста рецептора IL-1 [1].
Действие IL-1 и TNF-α на интенсивность резорбции костной ткани по крайней мере частично реализуется за счет стимулирования продукции простагландинов плазматическими клетками и гистиоцитами стенки радикулярной кисты, а также макрофагами или эндотелиальными клетками пульпы зуба. В частности, установлено, что IL-1 является индуктором транскрипции циклооксигеназы второго типа — фермента, обеспечивающего трансформацию арахидоновой кислоты в простагландины [12, 45, 70, 73]. Кроме того, имеются сведения, что IL-1 через воздействие на метаболизм тирозинкиназы регулирует цитоскелетную реорганизацию остеокластов, которая требуется для их активации [46].
В культуре тканей IL-1 (IL-1β) и трансформирующий фактор роста β (TGF-β) снижают экспрессию десневыми фибробластами матричной РНК тканевого ингибитора металлопротеиназы [TIMP-1 (tissue inhibitors of metallo-proteinase) mRNA] — ключевого фермента, обеспечивающего баланс между физиологической деструкцией экстраклеточного матрикса костной ткани и его синтезом [84]. Эти данные подтверждаются результатами, полученными в экспериментах in vivo. Выявлено, что IL-1α и TNFα, выделяемые макрофагами под действием LPS, стимулируют продукцию металлопротеиназы 1 [18].
Кроме того, как указывалось выше, IL1-β и TNFα, связываясь со специфическими мембранными рецепторами остеобластов и Т-лимфоцитов, могут стимулировать образование остеокластов опосредованно через повышение экспрессии RANKL [11, 17, 22]. При этом фактор роста опухолей-α индуцирует образование остеокластов через активацию одновременно двух факторов транскрипции — АР-1 и NFkB [32, 86].
Следует обратить внимание, что степень участия двух изоформ IL-1 (IL-1α и IL-1β) в стимулировании резорбции костной ткани не равнозначная. У человека уровень IL-1β в периапикальном экссудате в два раза выше, чем IL-1α [42]. В период обострения хронического апикального периодонтита (при наличии клинических симптомов заболевания) содержание IL-1β возрастает, а после лечения, наоборот, снижается на фоне увеличения уровня IL-1α [64]. У грызунов в составе экспериментально воспроизведенного очага периапикального воспаления преобладает IL-1 α.
IL-6 является плейотропным цитокином, способным в зависимости от обстоятельств усиливать или подавлять деструкцию костной ткани. Его продукция осуществляется клетками костной ткани под воздействием IL-1 и TNF — первичных медиаторов костной резорбции [4], а также паратгормона, родственного паратиреоидному гормону пептида и 1,25-дигидроксивитамина D [40]. В свою очередь IL-6 стимулирует ранние стадии гемопоэза, образование клеток-предшественников остеокластов в культуре тканей [36] и может увеличивать количество остеокластов in vivo, что в конечном итоге приводит к системному увеличению резорбции костной ткани.
Доказано присутствие IL-6 в неповрежденной периодонтальной связке и костной ткани, а также в инфицированной пульпе зуба экспериментальных животных [28]. Многократное превышение уровня IL-6 по сравнению с не инфицированной пульпой обнаружено в составе периапикальных очагов воспаления у человека [6, 8, 70]. По данным O. Takeishi et al. [70], IL-6 секретируют в очаг воспаления макрофаги, Т-лимфоциты, остеобласты или фибробласты. Высокое содержание IL-6 в составе периапикальных очагов воспаления дает основание предположить, что этот цитокин обладает остеолитическим эффектом [16]. Однако на основании данных, представленных в работах [4, 20], можно сделать противоположное заключение. Авторы установили, что у мутантных мышей с генетически детерминированным дефицитом IL-6 крупные очаги резорбции костной ткани в периапикальной области, индуцированные инфицированием пульпы зуба анаэробными бактериями, появляются значительно быстрее, чем у животных, не имеющих генетических дефектов. K. Balto et al. также показали, что при нейтрализации эндогенного IL-6 введением мышам анти-IL-6 антител выраженность костной резорбции существенно возрастала [47].
В ряде работ показано, что IL-6 подавляет индуцированную липополисахаридами продукцию IL-1 и TNF в моноцитах [2, 36, 57], а мыши с дефицитом IL-6 продуцируют в три раза больше TNF в ответ на стимуляцию LPS, чем экспериментальные животные без генетических дефектов. Приведенные данные дают авторам основание сделать заключение, что при воспалении, индуцированном эндотоксином, у IL-6 преобладают противовоспалительные свойства.
Вместе с тем нельзя исключать опосредованного влияния IL-6 на периапикальную резорбцию костной ткани через стимуляцию пролиферации эпителиальных клеток в составе радикулярной кисты [5]. В результате повышается гидростатическое давление внутри полости кисты и одновременно усиливается давление на окружающую костную ткань, стимулирующее ее резорбцию.
Кроме IL-6 в жидкости из полости радикулярной кисты и в тканях ее стенки обнаруживается высокое содержание других цитокинов, в частности GM-CSF и IL-3 [16]. Авторы [16] полагают, что эти цитокины могут участвовать в костной резорбции и прогрессивном увеличении размеров кисты. Однако данное утверждение не согласуется с результатами, полученными другими исследователями. Например, K. Shinoda et al. [58] показали, что в культуре тканей подавление дифференцировки остеокластов, индуцированное Т-лимфоцитами (именно эти клетки продуцируют GM-CSF — фактор, стимулирующий образование колоний гранулоцитов/макрофагов), частично нейтрализуется антителами к этому цитокину.
IL-2 и IFN-γ, которые продуцируются Th1 клетками (одна из субпопуляций СВ4-позитивных Т-лимфоцитов) в разгар периапикального воспаления, усиливают экспрессию IL-1, а цитокины Th2 клеток (IL-4, IL-5, IL-10), наоборот, угнетают продукцию IL-1 и других Th-1 цитокинов. H. Sasaki et al. установили, что главным супрессором костной резорбции in vivo является IL-10, эффект которого реализуется через подавление экспрессии IL-1α [53, 54]. Кроме того, остеокластогенез подавляют IL-4, а также IL-13 — цитокины, продуцируемые макрофагами [60].
Имеются публикации, в которых показано, что IL-12 и IL-18 индуцируют синтез IFN-γ и фактора, стимулирующего образование колоний гранулоцит-макрофагов (GM-CSF), которые в свою очередь подавляют остеокластогенез, по крайне мере в экспериментах in vitro [75, 76]. Мишенью для IFN-γ является TRAF 6 — вторичный посредник пути внутриклеточной передачи сигнала от RANK-рецепторов, который под действием цитокина разрушается [69]. Вместе с тем имеются сведения, что GM-CSF способен стимулировать образование остеокластов человека в культуре тканей благодаря активации в моноцитах синтеза фактора, активирующего образование колоний макрофагов (M-CSF) [15]. Хотя при высоких концентрациях GM-CSF (в присутствии RANKL и M-CSF) авторами регистрировалось торможение процесса образования остеокластов.
В последние годы появились сообщения о том, что эндогенные IL-12, IL-18 имеют статистически определенное значение в патогенезе резорбции костной ткани, индуцированной патогенными микроорганизмами in vivo [11, 53]. В частности, под влиянием IL-18 Т-лимфоциты, полученные из синовиальной ткани больных ревматоидным артритом, продуцируют RANKL.
Ингибирующим действием на остеокластогенез обладают инсулиноподобные факторы роста (insulin-like growth factors, IGF’s), которые синтезируются фибробластами и остеобластами. Низкий уровень IGF’s в сыворотке крови — фактор риска развития переломов костей [59]. При избытке белка, связывающего инсулиноподобный фактор роста (IGF-binding protein-3, IGFBP-3), наблюдается нарушение образования костной ткани и отмечается увеличение количества остеокластов.
Таким образом, во время обострения хронического воспалительного процесса вокруг верхушки корня зуба под действием бактериальных токсинов, проникающих в периапикальные ткани через апикальное отверстие, нарушается динамическое равновесие между процессами физиологического разрушения костной ткани и ее новообразования. Компоненты клеточной оболочки микроорганизмов оказывают прямое действие на остеобласты и преостеокласты, стимулируя продукцию сигнальных молекул, способствующих дифференцировке клеток, разрушающих костную ткань. Одновременно нарушаются регуляторные механизмы, сдерживающие этот процесс в физиологических условиях. Кроме того, продукты распада бактерий стимулируют экспрессию клетками, входящими в состав периапикального очага воспаления, остеолитических цитокинов, которые стимулируют дополнительную продукцию сигнальных молекул, под действием которых происходит морфологическая дифференцировка остеокластов и обеспечивается длительное поддержание их высокой функциональной активности.
Дальнейшее изучение молекулярных механизмов патогенеза резорбции костной ткани, опосредованной цитокинами, при локальных воспалительных процессах важно для лучшего понимания базовых механизмов функционирования остеокластов в физиологических условиях и при ряде патологических процессов, не связанных с воспалением. Известно, например, что давление на костную ткань, возникающее при окклюзионных и ортодонтических нагрузках, стимулирует ее локальное разрушение, а удаление зуба всегда сопровождается резорбцией альвеолярного гребня.
Универсальные внеклеточные и внутриклеточные сигнальные молекулы, обеспечивающие дифференцировку клеток-предшественников остеокластов и активное функционирование самих остеокластов, могут служить мишенью для новых лекарственных препаратов, основным эффектом которых должно стать снижение интенсивности локальной и генерализованной резорбции костной ткани.
В качестве примера препаратов, подавляющих процессы разрушения кости в составе челюстей, может служить иломостат (ilomostat) — ингибитор матричных металлопротеаз. Как указывалось выше, IL-1 (IL-1β) и трансформирующий фактор роста β (TGF-β), участвуют в регуляции продукции этих ферментов, которые интенсивно разрушают межклеточное вещество костной ткани.
Недавно было установлено, что партенолид (partenolide, PAR), содержащийся в лекарственных травах, останавливает резорбцию костной ткани (при маргинальном периодонтите), индуцированную липополисахаридами [85]. В основе его действия лежит супрессия ядерного фактора транскрипции каппа Б (NFkB), которая сопровождается угнетением остеокластогенеза и активацией апоптоза остеокластов. Одна из субъединиц NFkB, получившая название р65 пептида, может селективно ингибировать активность ядерного фактора транскрипции каппа Б, индуцированную первичным медиатором костной резорбции — фактором некроза опухолей [66].
Литература
1. Abramson S.B., Amin A. // Rheumatology (Oxford). 2002. V. 41, N 9. P. 972—980.
2. Aderka D.J., Le J., Wallach D. // J. Immunol. 1989. V. 143, N 11. P. 3517—3523.
3. Aubin J.E., Bonnelye E. // Osteoporos Int. 2000. V. 11, N 11. P. 905—913.
4. Balto K., White R., Mueller R., Stashenko P. // Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2002. V. 93, N 4. P. 461—468.
5. Bando Y., Henderson B., Meghji S., Poole S. // J. Oral Pathol. Med. 1993. V. 22, N 5. P. 221—227.
6. Barkhordar R.A., Hayashi C., Hussain M.Z. // Endod. Dent. Traumatol. 1999. V. 15, N 1. P. 26—27.
7. Bezerra M.C., Carvalho J.F., Prokopowitsch A.S., Pereira R.M. // Braz. J. Med. Biol. Res. 2005. V. 38, N 2. P. 161—170.
8. Bletsa A., Heyeraas K.J., Haug S.R., Berggreen E. // Neuroimmunomodulation. 2004. V. 11, N 6. P. 376—384.
9. Chikazu D., Katagiri M., Ogasawara T. et al. // J. Bone Miner. Res. 2001. V. 16, N 11. P. 2074—2081.
10. Corisdeo S., Gyda M., Zaidi M. et al. // Biochem. Biophys. Res. Commun. 2001. V. 285, N 2. P. 335—339.
11. Dai S.M., Nishioka K., Yudoh K. // Ann. Rheum. Dis. 2004. V. 63, N 11. P. 1379—1386.
12. Dinarello C.A. // Clin. Exp. Rheumatol. 2002. V. 20, N 5. Suppl. 27. P. 1—13.
13. Fouad A.F. // J. Dent. Res. 1997. V. 76, N 9. P. 1548—1554.
14. Fukushima H., Jimi E., Okamoto F. et al. // Bone. 2005. V. 36, N 2. P. 267—275.
15. Fujikawa Y., Sabokbar A., NealeS.D. et al. // Bone. 2001. V. 28, N 3. P. 261—267.
16. Gervбsio A.M., Silva D.A.O., Taketomi E.A. et al. // J. Dent. Res. 2002. V. 81, N 1. P. 64—66.
17. Hofbauer L.C., Lacey D.L., Dunstan C.R. et al. // Bone. 1999. V. 25, N 30. P. 255—259.
18. Hong C.Y., Lin S.K., Kok S.H. et al. // J. Oral. Pathol. Med. 2004. V. 33, N 3. P. 162—169.
19. Hou L., Sasaki H., Stashenko P. // Infect. Immun. 2000. V. 68, N 8. P. 4681—4687.
20. Huang G.T., Do M., Wingard M. et al. // Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2001. V. 92, N 1. P. 83—88.
21. Ikeda F., Nishimura R., Matsubara T. et al. // J. Clin. Invest. 2004. V. 114, N 4. P. 475—484.
22. Jiang Y., Mehta C.K., Hsu T.Y. // Infect. Immun. 2002. V. 70, N 6. P. 3143—3148.
23. Kabashima H., Yoneda M., Nakamuta H. et al. // J. Endod. 2004. V. 30, N 9. P. 634—657.
24. Kadono Y., Akiyama T., Ogata T. et al. // JBMR. 2001. V. 16. Suppl. 1. P. 176.
25. Kakehashi S., Stanley H.R., Fitzerald R.J. // Oral Surg. Oral Med. Oral Pathol. 1965. V. 20. P. 340—349.
26. Kanematsu M., Yoshimura K., Takaoki M., Sato A. // Bone. 2002. V. 30, N 4. P. 553—558.
27. Karst M., Gorny G., Galvin R.J., Oursler M.J. // J. Cell. Physiol. 2004. V 200, N 1. P. 99—106.
28. Kawashima N., Stashenko P. // Arch. Oral. Biol. 1998. V. 44, N 1. P. 55—66.
29. Kikuchi T., Matsuguchi T., Tsuboi N. et al. // J. Immunol. 2001. V. 166, N 5. P. 3574—3579.
30. Klinman D.M., Yi A.K., Beaucage S.L. et al. // Proc. Nat. Acad. Science USA. 1996. V. 93, N 7. P. 2879—2883.
31. Kobayashi N., Kadono Y., Naito A. // EMBO J. 2001. V. 20, N 6. P. 1271—1280.
32. Komine M., Kukita A., Kukita T. et al. // Bone. 2001. V. 28, N 5. 474—483.
33. Krieg A.M. // Ann. Rev. Immunol. 2002. V. 20. P. 709—706.
34. Krieg A.M., Yi A.K., Matson S. et al. // Nature. 1995. V. 374, N 6522. P. 546—549.
35. Kudo O., Fujikawa Y., ItonagaI. // J. Pathol. 2002. V. 198, N 2. P. 220—227.
36. Kurihara N., Bertolini D., Akiyam Y., Roodman G.D. // J. Immunol. 1990. V. 144, N 11. P. 4226—4230.
37. Lagasse E., Weissman I.L. // Cell. 1997. V. 89, N 7. P. 1021—1031.
38. Lam J., Takeshita S., Barjer J.E. et al. // Bone. 2001. V. 28, N 5. S1. OR 8.
39. Li X., Pilbeam C.C., Pan L. et al. // Bone. 2002. V. 30, N 4. P. 567—573.
40. Manolagas S.C., Jilka R.L. // New Engl. J. Med. 1995. V. 332, N. 5. P. 305—311.
41. Mбrton I.J., Kiss C. // Intern. Endod. J. 1993. V. 26, N 2. P. 131—136.
42. Matsuo T., Ebisu S., Nakanishi T. et al. // J. Endod. 1994. V. 20, N 9. P. 432—435.
43. Mei L.X., Liu Z., Zhang B. // Shanghai Kou Qiang Yi Xue. 2001. V. 10, N 1. P. 16—18.
44. Nair S.P., Meghji S., Wilson M. et al. // Infect. Immun. 1996. V. 64, N 7. P. 2371—2380.
45. Nakagawa T., Fujita N., Oh-Hara T. et al. // J. Cell. Physiol. 1999. V. 179, N 2. P. 226—232.
46. Nakamura I., Kadono Y., Takayanagi H. // J. Immunol. 2002. V. 168, N 10. P. 5103—5109.
47. Nakashima T., Kobayashi Y., Yamasaki S. et al. // Biochem. Biophys. Res. Commun. 2000. V. 275, N 3. P. 768—775.
48. Nukaga J., Kobayashi M., Shinki T. et al. // J. Periodontol. 2004. V. 75, N 2. P. 249—259.
49. Radics T., Kiss C., TarI., Marton I.J. // Oral Microbiol. Immunol. 2003. V. 18, N 1. P. 9—13.
50. Roux S., Orcel P. //Arthritis Res. 2000. V 2, N 6. P. 451—456.
51. Roux S., Orcel P. // Arthritis Res. 2000. V 2, N 6. P. 451—456.
52. Sabeti M., Simon J., Kermani V. et al. // J. Endod. 2005. V. 31, N 1. P. 17—18.
53. Sasaki H., Hou L., Belani A. et al. // J. Immunol. 2000. V. 165, N 7. P. 3626—3630.
54. Sasaki H., Balto K., Kawashima N. et al. // Clin. Diagn. Lab. Immunol. 2004. V 11, N 1. P. 106—110.
55. Shimauchi H., Takayama S., Imai-Tanaka T., Okada H. // J. Endod. 1998. V. 24, N 2. P. 116—119.
56. Shimauchi H., Takayama S., Narikawa-Kiji M. et al. // J. Endod. 2001. V. 27, N 12. P. 749—752.
57. Schindler R., Mancilla J., Endres S. et al. // Blood. 1990. V. 75, N 1. P. 40—47.
58. Shinoda K., Sugiyama E., Taki H. et al. // Bone. 2003. V. 33, N 4. P. 711—720.
59. Silha J.V., Mishra S., Rosen C.J. et al. // J. Bone. Miner. Res. 2003. V. 18, N 10. P. 1834—1841.
60. Simon L.S. The coexistence of osteoporosis and rheumatoid arthritis: pathophysiology. The role of IL-1 in bone resorption. The France Fondation. 2001. P. 24—28.
61. Sodek J., McKee M.D. // Periodontology. 2000. V. 24, N 1. P. 99—126.
62. Sommerfeldt D.W., Rubin C.T. // Eur. Spine J. 2001. V. 10 (Suppl. 2). P. 86—95.
63. Stashenko P., Wang C.Y., Tani-Ishii N., Yu S.M. // Oral Surg. Oral Med. Oral Pathol. 1994. V. 78, N 4. P. 494—502.
64. Stashenko P., Teles R., D’Souza. // Crit. Rew. Oral Biol. Med. 1998. V. 9, N 4. P. 498—521.
65. Strand V., Kavanaugh A.F. // Rheumatology (Oxford). 2004. V. 43. Suppl. 3. P. 10—16.
66. Takada Y., Singh S., Aggarwal B.B. // J. Biol. Chem. 2004. V. 279, N 15. P. 15096—15104.
67. Takahashi K. // Intern. Endod. J. 1998. V. 31, N 5. P. 311—325.
68. Takayama S., Miki Y., Shimauchi H., Okada H. // J. Endod. 1996. V. 22, N 12. P. 677—680.
69. Takayanagi H., Kim S., Taniguchi T. et al. // Arthritis. Res. 2002. N 4. Suppl. 3. S. 227—232.
70. Takeichi O., SaitoI., Tsurumachi T. // Calcif. Tissue. Intern. 1996. V. 58, N 4. P. 244—248.
71. Tani-Ishii N., Wang C.Y., Stashenko P. // Oral Microbiol. Immunol. 1995. V. 10, N 4. P. 213—219.
72. Tani-Ishii N., Tsunoda A., Teranaka T., Umemoto T. // J. Dent. Res. 1999. V. 78, N 10. P. 1617—162.
73. Tetradis S., Pilbeam C.C., Liu Y. et al. // Endocrinology. 1997. V. 138, N 9. P. 3594—3600.
74. Troen B.R. // Experimental. Gerontology. 2003. V. 38, N 6. P. 605—614.
75. Udagawa N. // J. Bone Miner. Metab. 2003. V. 21, N 6. P. 337—343.
76. Udagawa N., Kotake S., Kamatani N. // Arthritis. Res. 2002. V. 4, N 5. P. 281—289.
77. Vernal R., Chaparro A., Graumann R. et al. // J. Periodontol. 2004. V. 75, N 12. P. 1586—1591.
78. Viereck V., Emons G., Lauck V. et al. // Biochem. Biophys. Res. Commun. 2002. V. 291, N 3. P. 680—686.
79. Wada N., Maeda H., Tanabe K. et al. // J. Periodontal. Res. 2001. V. 36, N 1. P. 56—63.
80. Walker K.F., Lappin D.F., Takahashi K. et al. // Eur. J. Oral Sci. 2000. V. 108, N 3. P. 195—201.
81. Wilson M., Reddi K., Henderson B. // J. Periodontol. Res. 1996. V. 31, N 6. P. 393—407.
82. Wong B.R., Besser D., Kim N. et al. // Mol. Cell. 1999. V. 4, N 6. P. 1041—1049.
83. Xu L.X., Kukita T., Nakano Y. et al. // Lab. Invest. 1996. V. 75, N 5. P. 677—687.
84. Yang Y.Y., Tsai H.F., Lu S.C. et al. // J. Endod. 2002. V. 28, N 12. — P. 803—805.
85. Yip K.H., Zheng M.H., Feng H.T. et al. //J. Bone Miner. Res. 2004. V. 19, N 11. P. 1905—1916.
86. Zhang Y.H., Heulsmann A., Tondravi M.M. et al. // J. Biol. Chem. 2001. V. 276, N 1. P. 563—568.
87. Zou W., Bar-Shavit Z. // J. Bone Miner. Res. (a) 2002. V. 17, N 7. P. 1211—1218.
88. Zou W., Schwartz H., Endres S. et al. // FASEB J.2002 (b). V. 16, N 3. P. 274—282.
Современная стоматология. – 2005. – №4. – С. 20-26.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.
Содержание »
Архив »
|
|