По современным представлениям, ожирение у детей представляет собой хроническое прогрессирующее нарушение обмена веществ, характеризующееся избыточным увеличением массы тела ребенка относительно его роста и изменением состава тела, присущего данному возрасту [25, 26, 54, 66].
Причины детского ожирения подразделяют на генетические, метаболические, гормональные и средовые, вызывающие поломку механизма регуляции энергетического баланса организма и развитие заболевания [11, 54, 58, 66].
Генетические факторы
Ожирение — заболевание с полигенным типом наследования [54, 66]. В настоящее время известно более 20 генов, которые определяют процессы, оказывающие влияние на регуляцию энергетического гомеостаза организма и участвующие в генезе избыточной массы тела и ожирения, в том числе [3]:
• митохондриальный ген, кодирующий разобщающий протеин;
• ген проопиомеланокортина;
• ген лептина и его рецептора;
• ген, кодирующий фактор некроза опухоли;
• ген гликогенсинтетазы;
• ген инсулинрецепторного субстрата;
• ген липопротеиновой липазы;
• ген карбоксипептидазы Е (fat-ген);
• ген рецептора типа 4 меланоцитостимулирующего гормона и др.
Известно свыше 430 генов-кандидатов, но их роль в развитии ожирения у детей полностью не доказана [3, 36, 53, 54].
В работе [3] представлены генетически модулируемые факторы ожирения:
• cвязанные с обменом питательных веществ (макронутриентов): уровень липолиза в жировой ткани, активность липопротеинлипазы в жировой и мышечной ткани, состав и окислительный потенциал мышечной ткани, содержание свободных жирных кислот и β-рецепторная активность жировой ткани, способность к окислению жиров и углеводов, индивидуальные вкусовые предпочтения, регуляция аппетита;
• расходование энергии: скорость основного обмена, пост-алиментарный термогенез, распределение питательных веществ, уровень спонтанной мышечной активности;
• гормональные факторы: чувствительность к инсулину, секреция гормона роста, лептин.
В большинстве случаев детского ожирения отмечается многофакторность этого заболевания [5, 54, 66]. Гены-кандидаты повышают риск возникновения избыточной массы тела у ребенка только при условии действия средовых факторов (питание и образ жизни) [45, 54].
Выявлено несколько редких моногенных форм ожирения. Они вызваны мутациями генов лептина, рецептора лептина, конвертазы-1 прогормона, рецептора 4 меланокортина и проопиомеланокортина (табл. 1) [5, 15, 20 — 23, 34, 35, 63]. Клинические проявления моногенных вариантов характеризуются ранним началом заболевания, быстропрогрессирующим течением, морбидным ожирением, гиперфагией, вторичным гипогонадизмом.
|
|
|
|
|
|
|
|
|
Прогрессирующее с первых лет жизни морбидное ожирение. Выраженная гиперфагия
|
Неопределяемый уровень лептина. Гиперинсулинемия
|
Гипогонадотропный гипогонадизм. Ускорение костного созревания
|
|
|
|
Прогрессирующее с первых лет жизни морбидное ожирение. Выраженная гиперфагия
|
Гиперлептинемия. Гипер-инсулинемия умеренная. Сниженный инсулиноподобный фактор роста-1
|
Гипогонадотропный гипогонадизм. Сниженные темпы роста. Третичный гипотиреоз. Ускорение костного созревания
|
|
|
|
Прогрессирующее с первых лет жизни морбидное ожирение. Гиперфагия
|
Низкое содержание адренокортикотропного гормона и кортизола
|
Гипогонадотропный гипогонадизм. Рыжие волосы. Возможна гипогликемия
|
Ген конвертазы-1 прогормона
|
|
Прогрессирующее с первых лет жизни морбидное ожирение. Гиперфагия
|
Выраженная гиперпроинсулинемия. Гипоинсулинемия. Повышение уровня проопиомеланокортина. Низкое содержание адренокортикотропного гормона и умеренная гипокортизолемия. Нормальный стимулированный уровень кортизола
|
Гипогонадотропный гипогонадизм. Нарушение толерантности к углеводам с эпизодами постпрандиальных гипогликемий
|
Ген рецептора 4–го типа к меланокортину
|
|
Прогрессирующее с первых лет жизни морбидное ожирение. Гиперфагия
|
|
Ускорение темпов роста или высокорослость. Часто ускорение костного созревания
|
Таблица 1. Моногенные формы ожирения (по О.В. Ремизову, 2002)
(АР – аутосомно-рецессивный тип наследования, АД – аутосомно-доминантный тип наследования)
Некоторые генетические синдромы, например Прадера – Вилли, Беквита – Видемана, обусловлены нарушением процессов геномного импритинга. Высказано предположение, что с процессом импритинга (INS-IGF2 локус) связано развитие некоторых форм ожирения в сочетании с сахарным диабетом 2 типа и синдромом поликистозных яичников [5]. Определены различные локусы хромосом (2, 7, 12, 13, 22 хромосомы), связанных с ожирением.
Средовые факторы
В развитии детского ожирения важное место занимают экзогенные, или средовые факторы, в частности семейные стереотипы питания и пищевые привычки с употреблением продуктов с высоким содержанием жира и (или) калорий, прием пищи в вечернее и ночное время, невысокий социоэкономический статус семьи [18, 54, 67]. Дополнительным источником высококалорийного питания ребенка может быть еда, приготовленная вне дома, например в школе, в гостях, ресторанах fast-food. Ежедневно треть американских детей в возрасте 4—18 лет употребляет продукты быстрого питания, что приводит к дополнительному увеличению их массы тела до 6 фунтов в год [56]. Вариантом употребления продуктов с высоким содержанием калорий является так называемое безнадзорное питание детей (после школьных занятий). У подростков проявление социальной активности может выражаться в избыточном количестве перекусов вне дома и приеме продуктов с высоким содержанием жира и калорий [54].
Практический интерес имеет определение взаимосвязи частоты ожирения детей и принадлежности их к различным социальным группам. По данным польских исследователей, наиболее низкий процент детей с избыточной массой тела отмечается в семьях неквалифицированных рабочих. Вместе с тем установлено, что дети, родившиеся в менее обеспеченных семьях, страдали ожирением с более раннего возраста. Потеря одного из родителей чаще способствовала развитию поздних форм ожирения у ребенка. Показано также, что во всех группах минимальный процент детей, страдающих ожирением, отмечается в семьях, состоящих из 6 и более человек [66].
Низкая физическая активность или отсутствие адекватной физической нагрузки, увеличение времени пребывания у телевизора или компьютера, неподвижные игры (шахматы и т.п.) способствуют увеличению массы тела ребенка [37, 40, 42, 48, 67]. Степень ожирения в 8,3 раза выше у детей, проводящих за телевизором или компьютером более 5 часов в сутки, по сравнению с теми, кто смотрит телевизор (занят компьютером) менее 2 часов в день [46]. Только 22% американских школьников имеют рекомендуемый уровень физической активности, а 25% ведут полностью сидячий образ жизни [10, 18, 59].
В популяционных исследованиях установлено, что ожирение развивается у 14% детей, родители которых имели нормальную массу тела. Если ожирением страдает один из родителей, избыточная масса тела у потомства отмечается в 30—60% случаев. При наличии ожирения у обоих родителей ожирение развивается у 80% детей [54, 66]. Эти данные демонстрируют важную, но не абсолютную роль генетического фактора в развитии детского ожирения.
При обследовании 5000 выросших в разных приемных семьях разнояйцевых близнецов, биологические родители которых страдали ожирением, выявлено более существенное влияние факторов окружающей среды и пищевых стереотипов на формирование избыточной массы тела у ребенка, чем наследственная предрасположенность [54].
Метаболические и гормональные факторы
Жировая ткань рассматривается как важный эндокринный, метаболически активный «орган» [18, 40, 44, 47]. Вещества, вырабатываемые жировой тканью, обладают разнообразными биологическими эффектами и влияют на выраженность процессов во многих тканях и системах непосредственно или через нейроэндокринные механизмы [18, 44, 47]. Синтезируемые адипоцитами вещества играют ведущую роль во взаимосвязи ожирения и сопутствующих заболеваний (табл. 2) [3, 18, 44].
|
|
|
Заболевания и факторы риска, сопутствующие ожирению
|
Ангиотензиноген, ангиотензин II
|
Регуляция артериального давления
|
|
|
|
Воспаление, иммунный ответ, дифференцировка клеток
|
Онкологические заболевания, дистрофически-дегенеративные заболевания суставов
|
Ингибитор активатора плазминогена-1
|
|
Ишемическая болезнь сердца, тромбозы
|
Инсулиноподобный ростовой фактор-1
|
Апоптоз, рост и пролиферация клеток
|
Онкологические заболевания, осложнения сахарного диабета
|
|
|
Аппетит, инсулинорезистентность
|
Ожирение, сахарный диабет 2 типа
|
|
|
Воспаление, гемостаз, фертильность
|
Дистрофически-дегенеративные заболевания суставов, тромбозы
|
|
|
Инсулинорезистентность, липолиз, атеросклероз
|
Ожирение, ишемическая болезнь сердца, сахарный диабет 2 типа
|
|
|
Инсулинорезистентность, апоптоз клеток, атеросклероз, липогенез
|
Онкологические заболевания, сахарный диабет 2 типа, ишемическая болезнь сердца, нарушение фертильности
|
|
|
|
Нарушение менструального цикла и фертильности, онкологические заболевания
|
|
|
Улучшение чувствительности к инсулину, воспаление, атеросклероз
|
Сахарный диабет 2 типа, ишемическая болезнь сердца
|
Таблица 2. Роль основных биологически активных веществ, вырабатываемых в жировой ткани [3]
Жировая ткань подразделяется на два типа: коричневую и белую. Основное значение белой жировой ткани заключается в депонировании жира и образовании свободных жирных кислот, секреции лептина. Функции коричневой жировой ткани (КЖТ) — термогенез и процессы липолиза. У новорожденных КЖТ составляет 1 — 2% массы тела, с возрастом ее количество уменьшается [3].
КЖТ важна в генезе как генетического, так и алиментарного ожирения у детей. Ее активация происходит при участии симпатической нервной системы через β3- адренергические рецепторы. В митохондриях адипоцитов КЖТ выявлен разобщающий протеин (UCP1), способствующий высвобождению энергии в виде тепла, но не участвующий в расходовании энергии в виде мышечной работы. От активности β-адренергических рецепторов зависит повышение выделения UCP1 [3].
Особенности адренергической иннервации адипоцитов обусловливают влияние на скорость процессов липолиза и липогенеза, определяя количество депонированных триглицеридов в адипоците.
Жировая ткань висцеральной области имеет высокую метаболическую активность, в ней происходят процессы липолиза и липогенеза. Среди гормонов, участвующих в регуляции липолиза, важную роль играют катехоламины и инсулин: катехоламины – посредством взаимосвязи с α- и β-адренорецепторами, инсулин – через специфические рецепторы. Адипоциты висцеральной жировой ткани имеют высокую плотность адренорецепторов, особенно β3 - типа, и низкую плотность α-адренорецепторов и рецепторов к инсулину [3, 18, 44].
Одним из наиболее частых метаболических нарушений при ожирении у детей является феномен инсулинорезистентности (ИР). Частота и выраженность ИР возрастают при увеличении массы жировой ткани, особенно в висцеральной области [47].
Генез ИР при ожирении гетерогенен по природе. В ее развитии участвуют генетические, гормональные факторы, гиподинамия, условия внутриутробного развития ребенка и т.п. Большое значение в формировании ИР имеет сама жировая ткань, особенно висцеральные адипоциты [1, 47, 49]. Прямая зависимость между степенью развития жировой ткани и выраженностью ИР объясняется теорией липотоксичности [50]. Особенности висцеральных адипоцитов определяют их высокую липолитическую активность и поступление большого количества свободных жирных кислот (СЖК) по воротной вене в печень. При этом СЖК препятствуют связыванию инсулина гепатоцитами, обусловливая развитие ИР на печеночном уровне, снижение поглощения инсулина печенью и развитие системной гиперинсулинемии. В свою очередь гиперинсулинемия, нарушая ауторегуляцию инсулиновых рецепторов, усиливает периферическую ИР. СЖК также подавляют тормозящее действие инсулина на глюконеогенез, способствуя увеличению продукции глюкозы печенью. В мышечной ткани СЖК, конкурируя с субстратом в цикле глюкоза — жирные кислоты, препятствуют утилизации глюкозы миоцитами, что также ведет к развитию гипергликемии и компенсаторной гиперинсулинемии [1, 50].
В развитии и прогрессировании ИР и метаболических нарушений при ожирении важную роль играют адипоцитокины, секретируемые жировой тканью [1, 47, 71]. В первую очередь это лептин — нейрогормональный медиатор, продукт гена ожирения, локализованного на 7 хромосоме [71, 72].
Лептин продуцируется адипоцитами путем отщепления сигнальной последовательности из 21 аминокислоты от белков ob и состоит из 146 аминокислотных остатков [72]. Рецепторы лептина находятся в периферических тканях, включая легкие, почки, печень, поджелудочную железу, надпочечники, яичники, гемопоэтические клетки и скелетные мышцы [8, 39, 72]. Это позволяет говорить о лептине не только как о циркулирующем факторе насыщения.
Действие лептина проявляется на уровне гипоталамуса, где он связывается с ob-рецепторами, вызывая активацию сигналов, которые тормозят прием пищи и повышают расход энергии [24, 52, 71].
Уровень циркулирующего лептина пропорционален количеству жировой массы ребенка [12, 38, 71]. Отрицательный энергетический баланс организма, вызванный ограничением потребления пищи или голоданием, ведет к уменьшению концентрации лептина, тогда как положительный, вызванный перееданием, повышает его уровень [71, 72].
Существует модель регуляции энергетического баланса организма посредством лептина с вовлечением гипоталамических анаболического (нейропептид Y) и катаболического (α–меланоцитостимулирующий гормон и кортикотропин рилизинг-гормон) путей (рис. 1) [24, 38, 71].
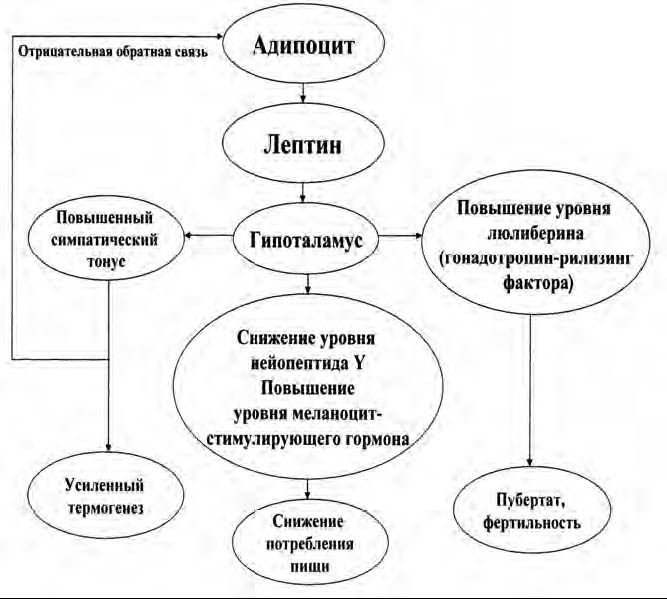
Рис. 1. Схема функционирования лептина
Пример анаболической системы — возрастание уровня нейропептида Y в аркуатном и паравентрикулярном ядрах гипоталамуса при отрицательном энергетическом балансе. Секреция нейропептида Y стимулирует аппетит, приводя к гиперфагии и гиперинсулинемии. Отмечается параллельная активация гипоталамус-гипофиз-надпочечниковой системы с увеличением выброса в кровь кортикостероидов [24, 71].
Гиперинсулинемия стимулирует накопление жировой ткани, в то время как повышенный уровень кортизола сдерживает утилизацию глюкозы организмом. Комбинация гиперинсулинемии и гиперкортизолемии является стимулом для продукции адипоцитами лептина. Повышенный уровень лептина вызывает увеличение уровня мРНК кортикотропин рилизинг-гормона и проопиомеланокортина/α-меланоцитостимулирующего гормона/ меланокортина в аркуатных ядрах гипоталамуса. Это ведет к уменьшению потребления пищи и снижению массы тела [71].
У детей уровень сывороточного лептина коррелирует с количеством общего, подкожного и висцерального жира, индексом массы тела. Изучены возрастные и половые особенности продукции лептина у детей [5, 6, 72]. В препубертате как у мальчиков, так и у девочек концентрация лептина в сыворотке крови низкая. В раннем пубертатном периоде (2-я стадия по Таннеру) отмечается увеличение уровня сывороточного лептина. После этого концентрация лептина снижается (установлена отрицательная корреляция с размерами яичек у мальчиков), достигая минимума на 5-й стадии развития гениталий. У девочек сывороточный уровень этого гормона остается постоянным до середины пубертата и достигает максимума на 5-й стадии по Таннеру [5, 6].
Дефицит лептина — достаточно редкое явление в детском возрасте. Сообщение о врожденном дефиците лептина у двух детей с выраженным ожирением вследствие мутации ob-рецептора, скорее, исключение [21]. У большинства детей с избыточной массой тела отмечается повышенный уровень лептина, что указывает на состояние лептинорезистентности или дисрегуляции [5, 6, 54]. Подкожное введение рекомбинантного лептина больным с ожирением подтвердило незначительный эффект в отношении снижения массы тела [26]. Лептинорезистентность может быть результатом как сигнального дефекта в лептинчувствительных нейронах гипоталамуса, так и нарушения транспорта гормона через гематоэнцефалический барьер [54].
Понимание физиологических механизмов регуляции потребления пищи, контроля аппетита и массы тела претерпело значительные изменения за последнее десятилетие [71]. Активно обсуждается роль в развитии ожирения эндоканнабиноидной системы (ЭС) [9]. ЭС обеспечивает контроль многих физиологических функций организма, включая регуляцию нервной и иммунной систем, энергетического обмена и репродукции, роста и дифференцировки клеток. Структура ЭС определена в процессе изучения биологических эффектов δ9-тетрагидроканнабинола – важнейшего компонента марихуаны и других дериватов растения Cannabis sativa (конопля посевная). ЭС включает каннабиноидные рецепторы КБ-1 и КБ-2, эндогенные каннабиноиды и ферменты, участвующие в процессе их биосинтеза. Рецепторы КБ-1 локализованы в центральной и периферической нервной системе, сердце, легких, эндотелии, желудочно-кишечном тракте, во всех органах эндокринной системы, а также в адипоцитах. Рецепторы КБ-2 находятся в клетках иммунной системы.
ЭС регулирует энергетический баланс в организме на основных функциональных уровнях, в которых задействованы лимбическая система, гипоталамус, желудочно-кишечный тракт, жировая ткань. Экзогенные и эндогенные каннабиноиды способствуют увеличению массы тела. При ожирении отмечается повышенная активность ЭС.
При ожирении наблюдается повышение выделения висцеральными адипоцитами фактора некроза опухолей–α. Этот цитокин обладает ауто- и паракринным действием и способствует развитию ИР преимущественно в жировой ткани [1, 18]. Под его влиянием снижается активность тирозинкиназы инсулинового рецептора, усиливается фосфорилирование серина — субстрата инсулинового рецептора и уменьшается экспрессия ГЛЮТ–4 (переносчик глюкозы–4) в мышечной и жировой ткани. Фактор некроза опухолей-α способствует развитию ИР также через стимуляцию липолиза в адипоцитах [1, 18].
Пропорционально нарастанию массы жировой ткани в крови увеличивается концентрация интерлейкина–6, который является ауто- и паракринным регулятором функции адипоцитов [1, 18, 40]. Этот адипоцитокин оказывает прямое воздействие на метаболические процессы в печени путем подавления в ней чувствительности рецепторов инсулина.
В жировой ткани секретируется адипонектин, участвующий в регуляции энергетического гомеостаза организма [51]. Плазменные концентрации адипонектина обратно коррелируют с выраженностью ожирения [29 — 31, 71]. Уровень адипонектина повышается при голодании и снижении массы тела на фоне низкокалорийной диеты у лиц с ожирением [29, 71]. Установлено, что низкий уровень адипонектина в крови предшествует развитию ИР [1, 30]. В эксперименте показано, что адипонектин уменьшает ИР, стимулируя фосфорилирование тирозина – рецептора инсулина. Кроме того, механизм влияния адипонектина на ИР заключается в снижении поступления жирных кислот в печень и в стимуляции их окисления путем активации протеинкиназы, что приводит к уменьшению продукции глюкозы печенью, а также к синтезу липопротеидов очень низкой плотности [1, 30, 44].
В генезе ожирения важная роль отводится резистину, участвующему в развитии ИР [55, 64], и грелину — пептидному гормону, секретируемому клетками гастроинтестинального тракта, влияющему на активацию орексигенных пептидов в гипоталамусе [32, 70].
Грелин – пептид, состоящий из 28 аминокислотных остатков, секретируется в желудке, двенадцатиперстной кишке и кишечнике [16, 49]. Это непосредственный стимулятор аппетита, регулирующий потребление пищи и энергетический баланс организма [13, 43, 68]. Он активирует нейроны гипоталамуса и аркуатных ядер, смежных с 3-м желудочком, что приводит к выбросу рилизинг-гормона соматотропного гормона, нейропептида Y, агонистов меланокортин-рецептора, агутипротеина и к положительному энергетическому балансу благодаря стимуляции потребления пищи и снижению утилизации жира [2]. Уровень грелина выше натощак и снижается после еды [7, 13, 61, 68], что позволяет предположить его роль в пищевом поведении [49, 60, 70]. Введение грелина стимулирует потребление пищи у крыс, повышает массу тела и уменьшает утилизацию жира [60, 69]. У лиц с ожирением внутривенная инфузия этого пептида вызывает увеличение количества принятой пищи на 28% [68]. Кроме того, у них выявлен ночной подъем уровня грелина, превышающий пики, ассоциированные с приемом пищи, что косвенно объясняет повышенный аппетит в вечернее и ночное время [2].
Уровни грелина натощак и постпрандиально не претерпевают изменений после краткосрочного ограничения калорийности, в отличие от значительного снижения уровня лептина [13, 57, 58]. Содержание пептида повышается только после уменьшения массы тела на фоне длительной диеты. Этим объясняется выраженное снижение массы тела у пациентов после резекции желудка по сравнению с лицами, соблюдающими строгую диету: после хирургического вмешательства уровень грелина уменьшается до неопределяемых значений, в то время как высокий уровень грелина на фоне строгой диеты приводит к повышению чувства голода [2].
Влияние грелина на обмен глюкозы и инсулин неоднозначно. С одной стороны, он стимулирует секрецию инсулина у животных как in vitro, так и in vivo, с другой — выявлено его ингибирующее воздействие на уровень инсулина, предварительно стимулированного глюкозой [2]. Это позволяет предположить, что постпрандиальное повышение уровня инсулина способствует снижению грелина. Уровень грелина ниже у больных с инсулинорезистентностью, нежели у пациентов с сопоставимым индексом массы тела и нормальной чувствительностью к инсулину [13].
Изменения гена грелина могут вызвать ожирение в раннем детском возрасте [33, 41] или служить защитным механизмом накопления жира [62], но окончательная роль полиморфизма грелина в контроле веса не уточнена [28, 65].
Заслуживает внимания изучение грелина у больных с синдромом Прадера-Вилли, который характеризуется повышенным аппетитом, прогрессирующим ожирением, задержкой умственного развития, низкорослостью в сочетании с гипогонадизмом, нарушением углеводного обмена. Выраженная гиперфагия при этом синдроме связана с повышенным уровнем грелина, причина которого не установлена [14, 23]. Ни грелин, ни его рецептор не относятся к определенному участку хромосомы 15q11-13, ответственной за данный синдром. Возможно, дальнейшее изучение данного феномена поможет объяснить патогенез ожирения при синдроме Прадера-Вилли [2, 14, 23].
Большое внимание уделяется изучению мутаций выделенного в жировой ткани РРАR-γ рецептора, связанного с обменом глюкозы и жира. РРАR-γ относится к факторам транскрипции и играет важную роль в процессе дифференцировки адипоцитов, участвует в экспрессии гена, отвечающего за синтез белка, транспортирующего жирные кислоты, тормозит экспрессию ob-гена и фактора некроза опухоли-α, регулирует выработку белков, разобщающих окислительное фосфорилирование [71].
Регуляция аппетита и энергетического гомеостаза
Влияние внешних факторов на регуляцию аппетита и энергетического баланса в организме реализуется через нервную и эндокринную системы (рис. 2) [18, 71].
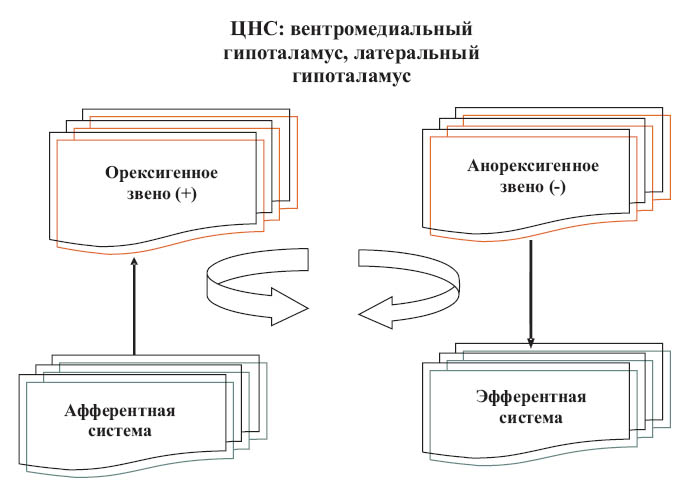
Рис.2. Модель регуляции аппетита и энергетического баланса в организме
Выделяют:
• афферентную систему, включающую лептин и другие короткодействующие факторы насыщения и пищевые сигналы (грелин, низкий уровень глюкозы плазмы, кортизол, белки, гормоны желудочно-кишечного тракта);
• эфферентную систему, представляющую собой комплекс факторов аппетита (насыщения), моторики и термогенеза;
• неэндокринную часть, представленную желудочно-кишечным трактом, ответственную за адсорбцию и метаболизм;
• центральную нервную систему с вентромедиальными, аркуатными и паравентрикулярными ядрами вентромедиального гипоталамуса и латеральным гипоталамусом.
Гипоталамус активно участвует в регуляции энергетического баланса и пищевого поведения ребенка. Повреждение вентромедиальных ядер приводит к постоянной гиперфагии и морбидному ожирению, а повреждение латерального гипоталамуса сопровождается потерей аппетита, адипсией и потерей массы тела. Гипоталамическая регуляция аппетита заключается во взаимодействии орексигенных и анорексигенных аминов и пептидов, связанных с афферентной и эфферентной системами [18, 71].
К орексигенным факторам относятся нейропептид Y, опиоидные пептиды, орексины А и Б, гипокретины, галанин, глютамат, норэпинефрин, меланокортикотропный гормон и др.
Анорексигенные факторы представлены проопиомеланокортином и α–меланоцитостимулирующим гормоном, рецепторами меланокортина, допамином, серотонином, нейротензином, кортикотропин рилизинг-гормоном, кокаин-регулируемым и амфетамин-регулируемым транскрипторами.
В регуляции термогенеза значительная роль отводится и другим, малоизученным механизмам, принимающим участие в контроле поступления и расхода энергии (экспрессия и полиморфизм α3- адренорецепторов, обмен трансретиноевой кислоты и др.) [71].
Исследование комплексной системы регуляции аппетита и энергетического гомеостаза, открытие и изучение новых аспектов развития ожирения способствуют развитию и совершенствованию терапии избыточной массы тела в детском возрасте. Используемые сегодня в педиатрической практике лекарственные препараты (сибутрамин, орлистат) имеют ограниченные показания и длительность терапии (1—2 года) [18, 27, 54]. Хирургические методы лечения ожирения у детей не нашли широкого применения. Окончательное уточнение механизмов нарушения энергетического гомеостаза позволит назначать эффективную индивидуально подобранную терапию, основанную на использовании периферических гормонов или нейромедиаторов. Примером такого успешного лечения является назначение детям препаратов рекомбинантного лептина при морбидном ожирении, вызванном дефицитом этого гормона [19]. Таким образом, адекватная терапевтическая стратегия может существенно снижать массу тела и влиять на эндокринные и метаболические осложнения ожирения, приводящие к увеличению заболеваемости и смертности у взрослых.
Литература
1. Бутрова С.А., Плохая А.А. // Сахарный диабет. – 2005. — № 3. – С.45—50.
2. Васюкова О.В., Витебская А.В. // Проблемы эндокринологии. – 2006. – Т. 52, № 2. – С.3—7.
3. Ивлева А.Я., Старостина Е.Г. Ожирение – проблема медицинская, а не косметическая. – М., 2002.
4. Ожирение / под ред. И.И. Дедова, Г.А. Мельниченко. – М., 2004. – С.312—328.
5. Солнцева А.В. // Актуальные вопросы эндокринологии: тез. докл.— СПб., 2000. – С.247.
6. Солнцева А.В. // Здравоохранение. – 2002. — № 1. – С. 6—9.
7. Ariyasu H., Takaya K., Tagami T. et al. // J. Clin. Endocrinol. Metab. — 2001. – Vol. 86. – P. 4753—4758.
8. Bado A., Levasseur S., Attoub S. et al. // Nature. — 1998. — Vol. 394. — P. 790–793.
9. Black S.C. // Curr. Opin. Investig. Drugs. – 2004. – Vol. 5. – P. 389–394.
10. Burgeson C.R., Wechsler H., Brener N.D. et al. // J. Sch. Health. — 2001. – Vol.71. – P. 279—293.
11. Clement K., Ferre P. // Pediatr. Res. – 2003. – Vol. 53. – P. 721–725.
12. Considine R.V., Sinha M.K., Heiman M.L. et al. // New Engl. J. Med. – 1996. — Vol. 334. – P. 292–295.
13. Cummings D.E., Purnell J.Q., Frayo R.S. et al. // Diabet. – 2001. – Vol. 50. – P. 1714–1719.
14. Cummings D.E., Clement K., Purnell J.Q. et al. // Nat. Med. – 2002. — Vol. 8. – P.643–644.
15. Damcott C.M., Feingold E., Moffett S.P. et al. // Metabolism. – 2004. – Vol. 53. – P. 458–464.
16. Date Y., Kojima M., Hosoda H. et al. // Endocrinol. – 2000. – Vol. 141. — P. 4255–4261.
17. Diamond Jr. F. B., Eichler D.C. // Crit. Rev. Clin. Labor. Sci. – 2002. – Vol. 39. – P. 499–525.
18. Druce M., Bloom S. R. // Arch. Dis. Child. – 2006. – Vol. 91. – P.183—187.
19. Farooqi I.S., Jebb S.A., Langmack G. et al. // New Engl. J. Med. – 1999. – Vol. 341. – P. 879–884.
20. Farooqi I.S., Yeo G.S., Keogh J.M. et al. // J. Clin. Invest. – 2000. – Vol. 106. – P. 271–279.
21. Farooqi I.S., Keogh J.M., Kamath S. et al. // Nature. – 2001. – Vol. 414. – P. 34–35.
22. Farooqi I.S., Matarese G., Lord G.M. et al. // J. Clin. Invest. – 2002. – Vol. 110. – P. 1093–1103.
23. Farooqi I.S., O’Rahilly S. // Annu. Rev. Med. – 2005. – Vol. 56. – P. 443—458.
24. Frederich R.C., Lollmann B., Hamann A. et al. // J. Clin. Invest. – 1995. — Vol. 96. – P. 1658–1663.
25. Freedman D.S., Khan L.K., Serdula M.K. et al. // Intern. J. Obes. Relat. Metab. Disord. – 2004. – Vol. 28. – P. 10–16.
26. Freemark M. // Diabetes Care. – 2007. – Vol.30. – P.395—402.
27. Grace C., Beales P., Summerbell C. et al. // Intern. J. Obes. Relat. Metab. Disord. – 2003. – Vol. 27. – P.1319–1324.
28. Hinney A., Hoch A., Geller F. et al. // J. Clin. Endocrinol. Metab. — 2002. – Vol. 87. – P. 2716–2719.
29. Hotta K., Funahashi T., Arita Y. et al. // Arterioscl., Thromb. Vascul. Biol. – 2000. – Vol. 20. – P. 1595–1599.
30. Hotta K., Funahashi T., Bodkin N.L. et al. // Diabet. – 2001. – Vol. 50. – P. 1126–1133.
31. Hu E., Liang P., Spiegelman B.M. // J. Biol. Chem. – 1996. – Vol. 271. – P. 10697–10703.
32. Kojima M., Hosoda H., Date Y. et al. // Nature. —1999. – Vol. 402. – P. 656–660.
33. Korbonits M., Gueorguiev M., O’Grady E. et al. // J. Clin. Endocrinol. Metab. — 2002. – Vol. 87. – P. 4005–4008.
34. Krude H., Biebermann H., Gruters A. // Ann. NY Acad. Sci. – 2003. – Vol. 994. – P. 233–239.
35. Krude H., Biebermann H., Schnabel D. et al. // J. Clin. Endocrinol. Metab. – 2003. – Vol. 88. – P. 4633–4640.
36. Le Stunff C., Fallin D., Schork N.J. et al. // Nat. Genet. – 2000. – Vol. 26. – P. 444—446.
37. Livingstone M.B., Robson P.J., Wallace J.M. et al. // Proc. Nutr. Soc. – 2003. – Vol. 62. – P. 681–701.
38. Maffei M., Halaas J., Ravussin E. et al. // Nat. Med. – 1995. – Vol.1. – P. 1155–1161.
39. Masuzaki H., Ogawa Y., Sagawa N. et al. // Nat. Med. – 1997. – Vol. 3. – P.1029–1033.
40. Miller J., Rosenbloom A., Silverstein J. // J. Clin. Endocrinol. Metab. — 2004. – Vol. 89. –P. 4211—4218.
41. Miragliad G., Santoro N., Cirillo G. et al. // Intern. J. Obes. Relat. Metab. Dis. — 2004. — Vol. 28. – P. 447—450.
42. Moore L.L., Gao D., Bradlee M.L. et al. // Prev. Med. – 2003. – Vol. 37. – P.10–17.
43. Nakazato M., Murakami N., Date Y. et al. // Nature. – 2001. – Vol. 409. – P. 194–198.
44. Ouchi N., Kihara S., Funahashi T. et al. // Curr. Opin. Lipidol. – 2003. – Vol. 14. – P. 561–566.
45. Perusse L., Bouchard C. // Amer. J. Clin. Nutr. – 2000. – Vol. 72. – P.1285S–1290S.
46. Proctor M.H., Moore L.L., Gao D. et al. // Intern. J. Obes. Relat. Metab. Disord. – 2003. – Vol. 27. – P. 827–833.
47. Rajala M.W., Scherer P.E. // Endocrinol. – 2003. – Vol. 144. – P. 3765–3773.
48. Saelens B.E., Sallis J.F., Nader P.R. et al. // J. Dev. Behav. Pediatr. – 2002. – Vol. 23. – P.127–132.
49. Sakata I., Nakamura K., Yamazaki M. // Peptid. – 2002. – Vol. 23. – P. 531–536.
50. Schaffer J.E. // Curr. Opin. Lipidol. – 2003. – Vol. 14. – P. 281–287.
51. Scherer P.E., Williams S., Fogliano M. et al. // J. Biol. Chem. – 1995. – Vol. 270. – P. 26746–26749.
52. Seeley R.J., Yagaloff K.A., Fisher S.L. et al. // Nature. – 1997. – Vol. 390. – P. 349.
53. Snyder E.E., Walts B., Perusse L. et al. // Obes. Res. – 2004. – Vol. 12. – P. 369–439.
54. Speiser P.W., Rudolf M.C.J., Anhalt H. et al. // J. Clin. Endocrinol. Metab. – 2005. — Vol. 90. — P. 1871—1887.
55. Steppan C.M., Bailey S.T., Bhat S. et al. // Nature. – 2001. – Vol. 409. – P. 307–312.
56. St-Onge M.P., Keller K.L., Heymsfield S.B. // Amer. J. Clin. Nutr. — 2003. —Vol. 78. – P. 1068—1073.
57. Sugino T., Yamaura J., Yamagishi M. et al. // Biochem. Biophys. Res. Commun. – 2002. — Vol. 298. – P. 785–788.
58. Sun Y., Ahmed S., Smith R.G. // Molecul. Cell. Biol. – 2003. – Vol. 23. – P. 7973–7981.
59. Troiano R.P. // New Engl. J. Med. – 2002. — Vol. 347. – P. 706–707.
60. Tschop M., Smiley D.L., Heiman M.L. // Nature. – 2000. – Vol. 407. – P. 908–913.
61. Tschop M., Wawarta R., Riepl R.L. et al. // J. Endocrinol. Invest. – 2001. – Vol. 24. — RC19–RC21.
62. Ukkola O., Ravussin E., Jacobson P. et al. // Obes. Res. – 2002. – Vol. 10. – P. 782–791.
63. Vaisse C., Clement K., Durand E. et al. // J. Clin. Invest. – 2000. – Vol. 106. – P. 253–262.
64. Valsamakis G., McTernan P.G., Chetty R. et al. // Metabolism. – 2004. – Vol. 53. – P. 430–434.
65. Wang H.J., Geller F., Dempfle A. et al. // J. Clin. Endocrinol. Metab. — 2004. – Vol. 89. –P. 157–162.
66. Wechsler J.G. Adipositas: Ursachen und Therapie. – Blackwell Verlag GmbH, Berlin, Wien, 2003.
67. Wilson N., Quigley R., Mansoor O. // J. Public. Health. — 1999. —Vol. 23. – P. 647–650.
68. Wren A.M., Small C.J., Ward H.L. et al. // Endocrinol. – 2000. – Vol. 141. – P. 4325—4328.
69. Wren A.M., Small C.J., Abbott C.R. et al. // Diabet. – 2001. – Vol. 50. – P. 2540–2547.
70. Wren A.M., Seal L.J., Cohen M.A. et al. // J. Clin. Endocrinol. Metab. — 2001. – Vol. 86. – P. 5992.
71. Wynne K., Stanley S., McGowan B. et al. // J. Endocrinol. – 2005. – Vol.184. – P.291—318.
72. Zhang Y., Proenca R., Maffei M. et al. // Nature. — 1994. – Vol. 372. – P. 425–432.
Медицинские новости. – 2008. – №3. – С. 7-13.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.